

The dynamics of molecular evolution over 60,000 generations
- PMID: 29045390
- PMCID: PMC5788700
- DOI: 10.1038/nature24287
The dynamics of molecular evolution over 60,000 generations
Abstract
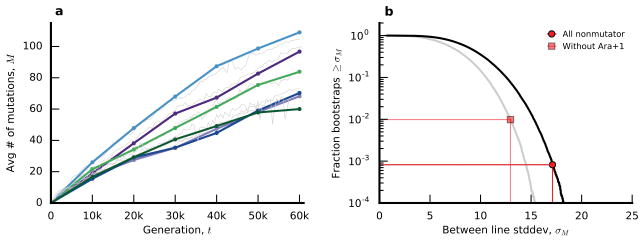
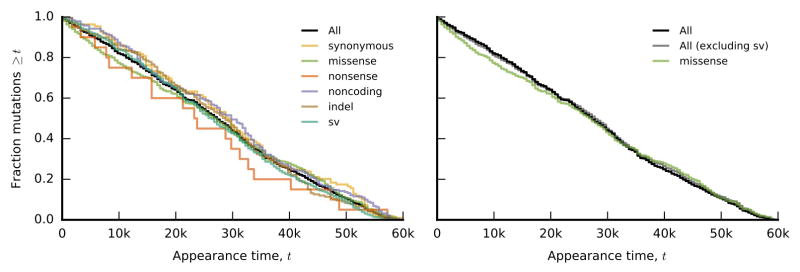
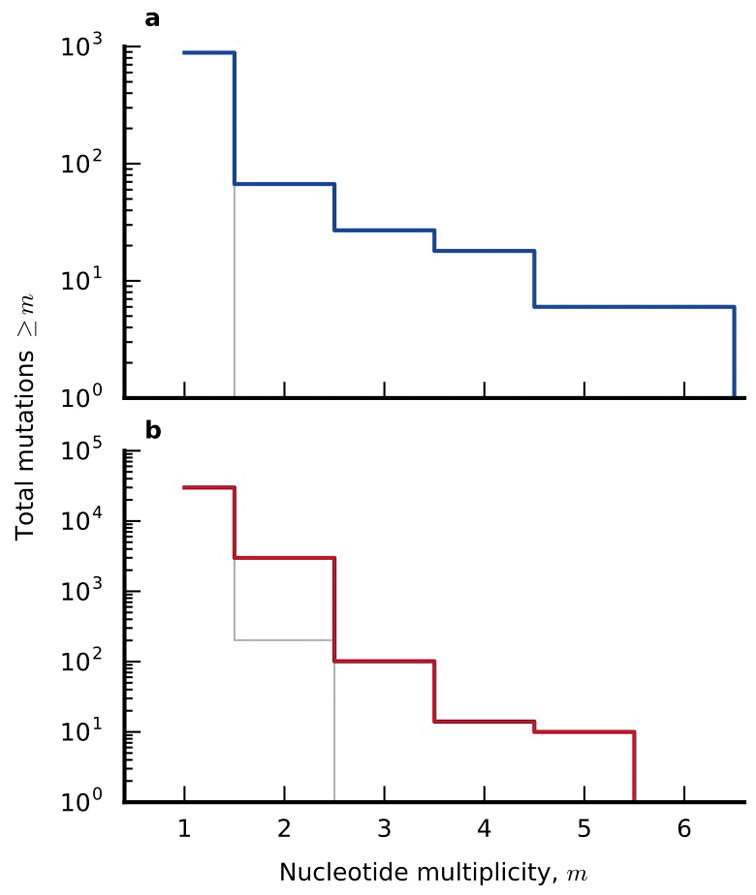
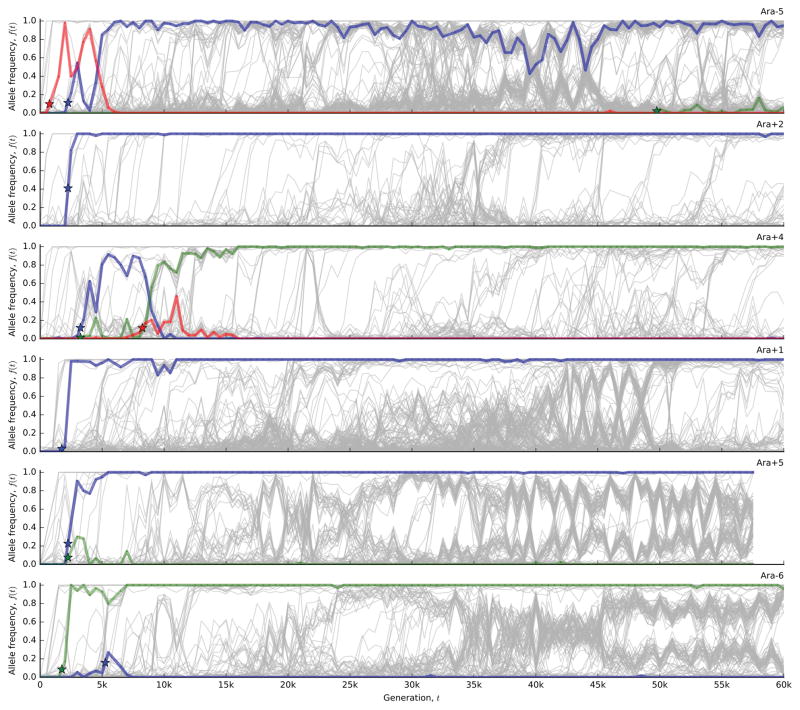
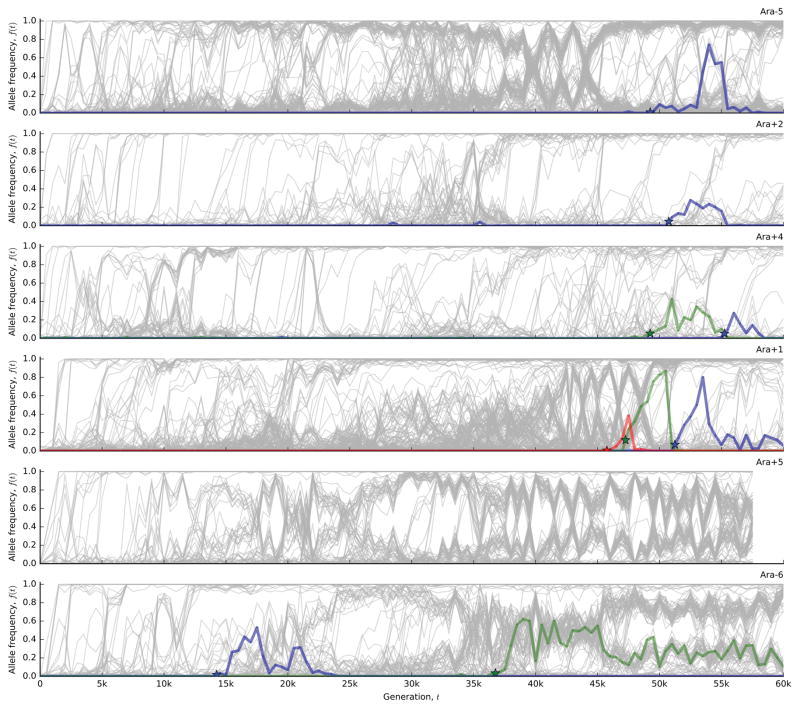
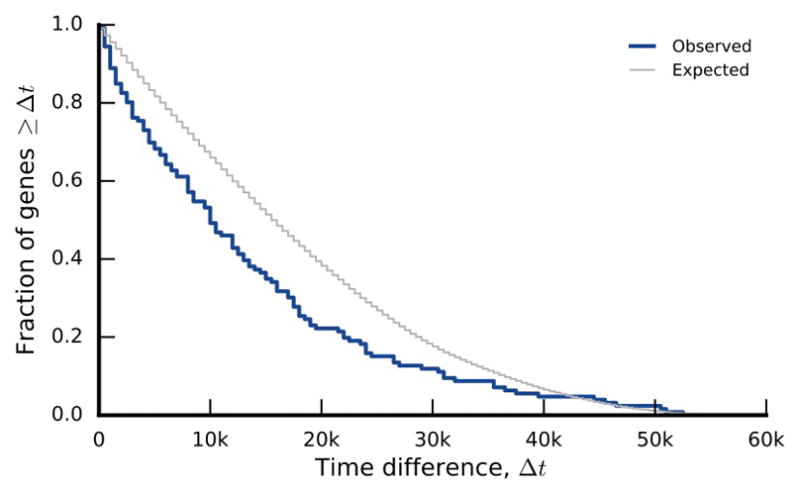
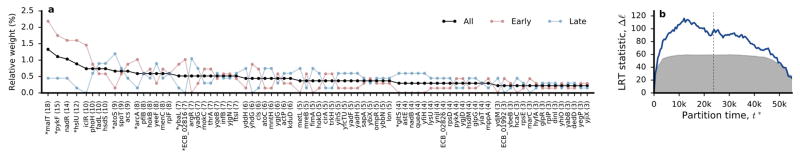
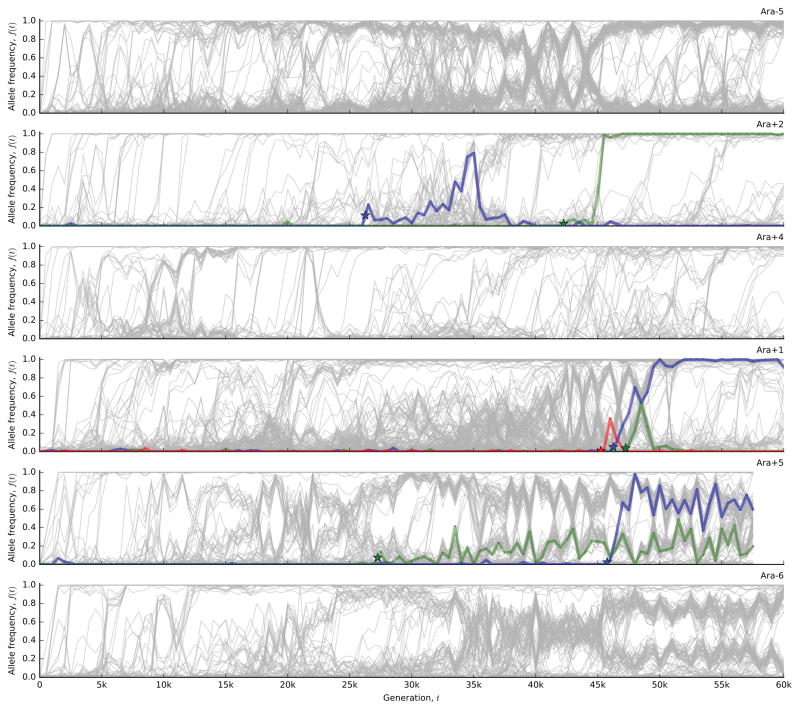
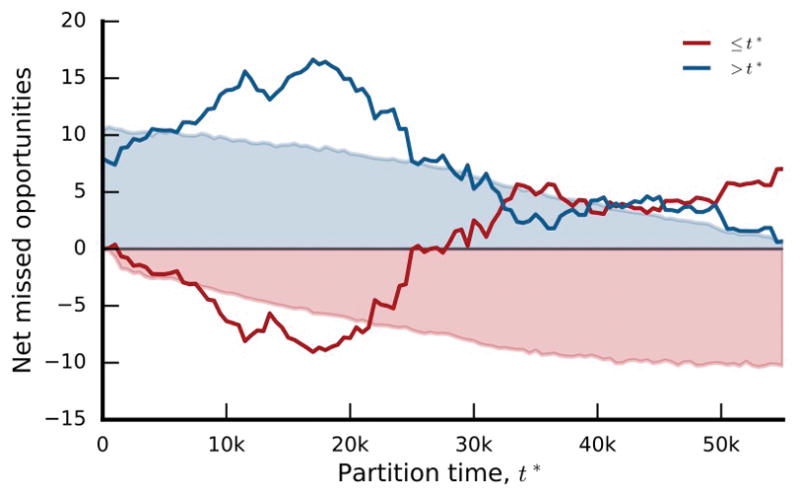
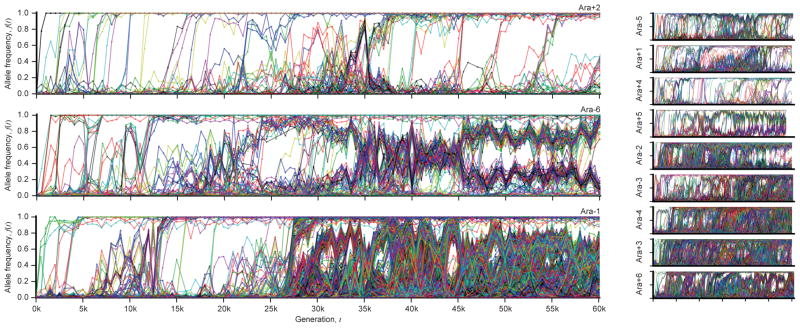
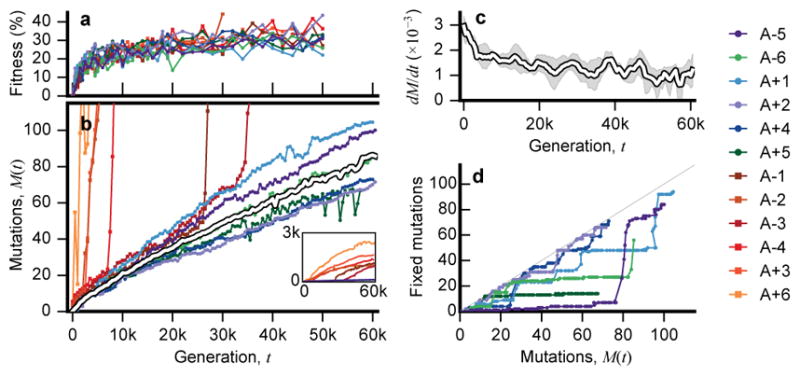
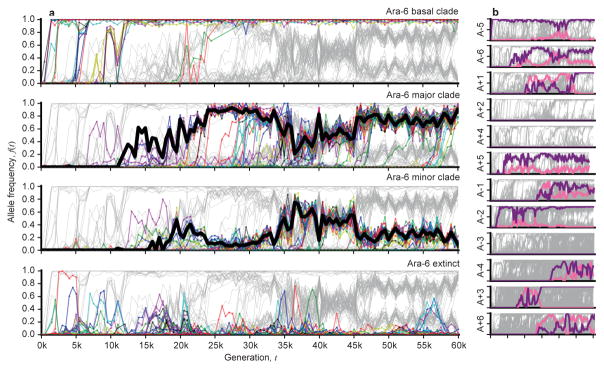
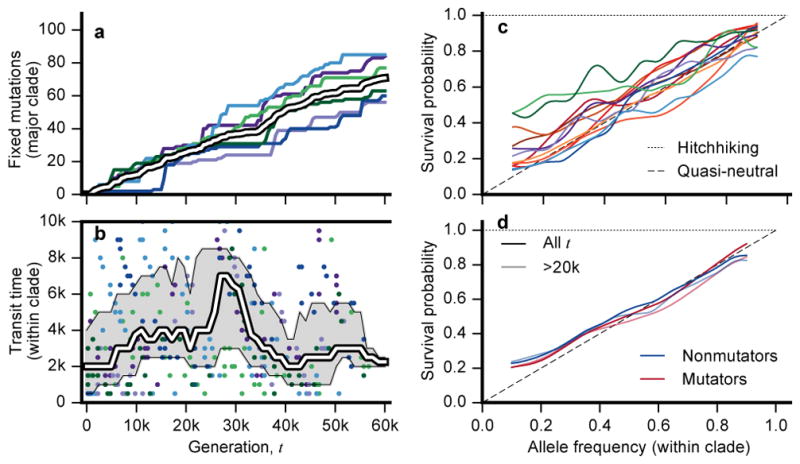
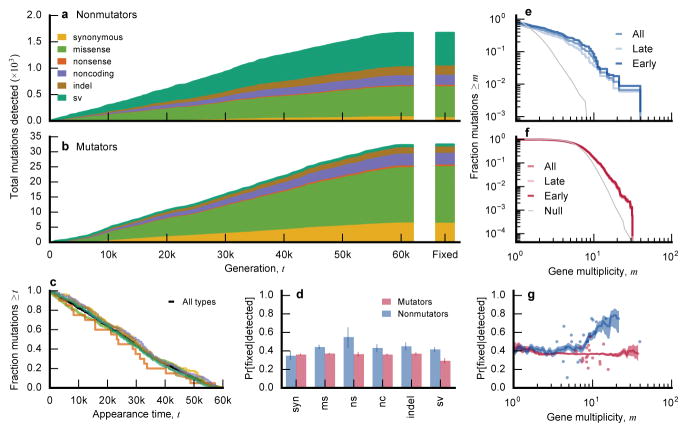
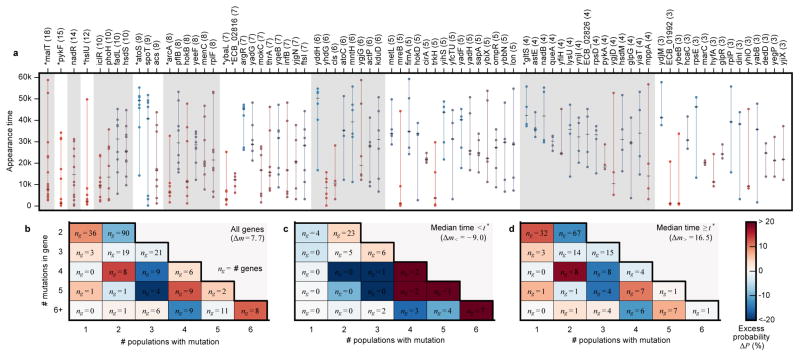
The outcomes of evolution are determined by a stochastic dynamical process that governs how mutations arise and spread through a population. However, it is difficult to observe these dynamics directly over long periods and across entire genomes. Here we analyse the dynamics of molecular evolution in twelve experimental populations of Escherichia coli, using whole-genome metagenomic sequencing at five hundred-generation intervals through sixty thousand generations. Although the rate of fitness gain declines over time, molecular evolution is characterized by signatures of rapid adaptation throughout the duration of the experiment, with multiple beneficial variants simultaneously competing for dominance in each population. Interactions between ecological and evolutionary processes play an important role, as long-term quasi-stable coexistence arises spontaneously in most populations, and evolution continues within each clade. We also present evidence that the targets of natural selection change over time, as epistasis and historical contingency alter the strength of selection on different genes. Together, these results show that long-term adaptation to a constant environment can be a more complex and dynamic process than is often assumed.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Molecular evolution: No escape from the tangled bank.Nature. 2017 Nov 2;551(7678):42-43. doi: 10.1038/nature24152. Epub 2017 Oct 18. Nature. 2017. PMID: 29045393 No abstract available.
References
-
- Barrick JE, et al. Genome evolution and adaptation in a long-term experiment with Escherichia Coli. Nature. 2009;461:1243–1247. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
