

Clonal Heterogeneity Influences the Fate of New Adaptive Mutations
- PMID: 29045840
- PMCID: PMC5656752
- DOI: 10.1016/j.celrep.2017.09.046
Clonal Heterogeneity Influences the Fate of New Adaptive Mutations
Abstract
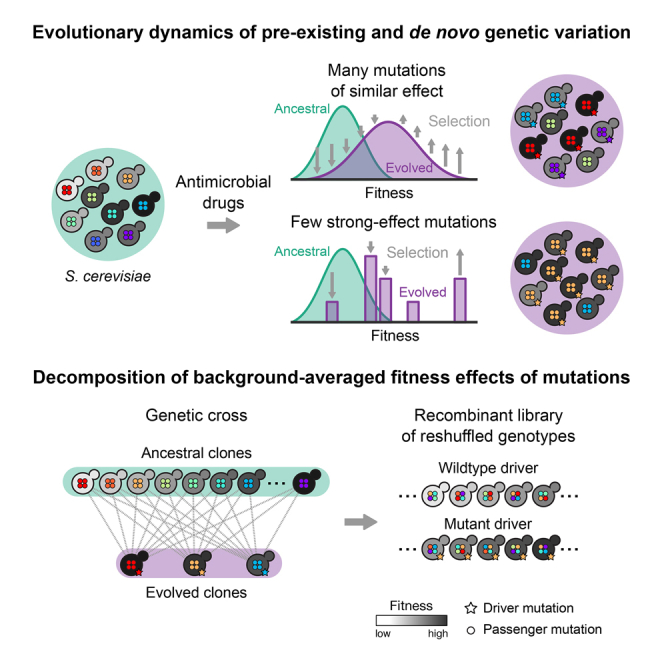
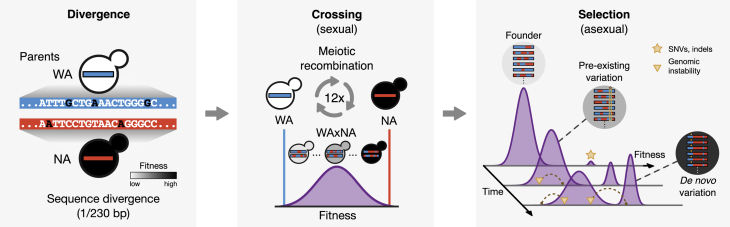
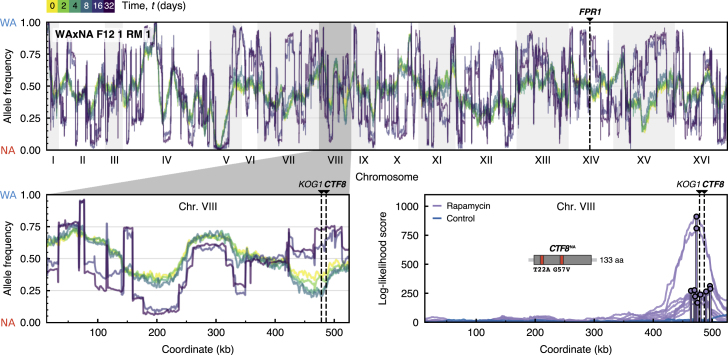
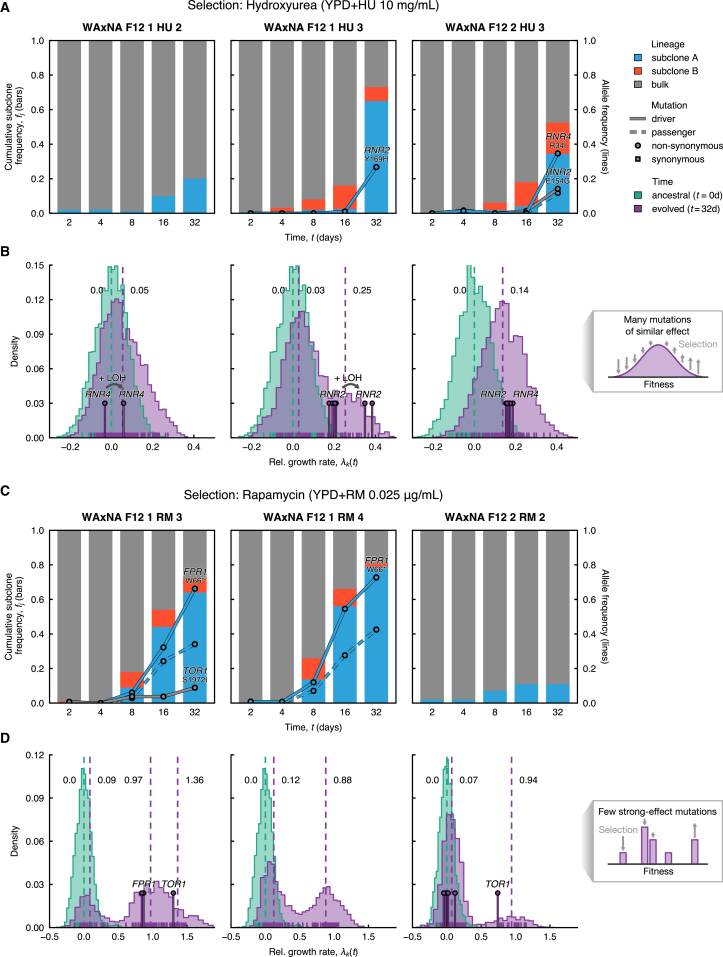
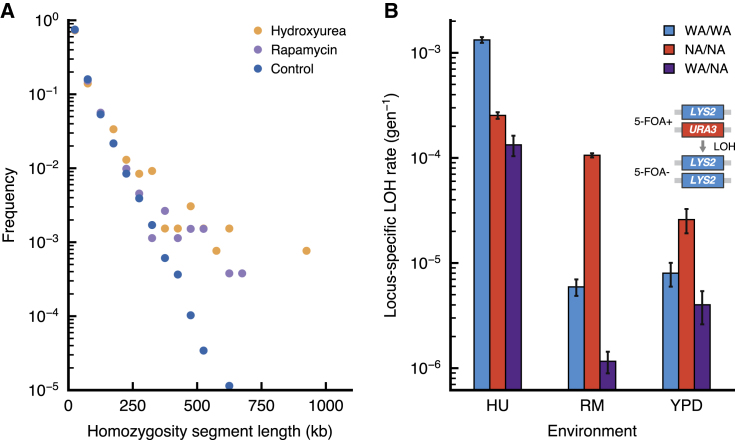
The joint contribution of pre-existing and de novo genetic variation to clonal adaptation is poorly understood but essential to designing successful antimicrobial or cancer therapies. To address this, we evolve genetically diverse populations of budding yeast, S. cerevisiae, consisting of diploid cells with unique haplotype combinations. We study the asexual evolution of these populations under selective inhibition with chemotherapeutic drugs by time-resolved whole-genome sequencing and phenotyping. All populations undergo clonal expansions driven by de novo mutations but remain genetically and phenotypically diverse. The clones exhibit widespread genomic instability, rendering recessive de novo mutations homozygous and refining pre-existing variation. Finally, we decompose the fitness contributions of pre-existing and de novo mutations by creating a large recombinant library of adaptive mutations in an ensemble of genetic backgrounds. Both pre-existing and de novo mutations substantially contribute to fitness, and the relative fitness of pre-existing variants sets a selective threshold for new adaptive mutations.
Keywords: adaptation; clonal heterogeneity; drug resistance; genetic variation; genome evolution; mutation; population dynamics; quantitative traits.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures

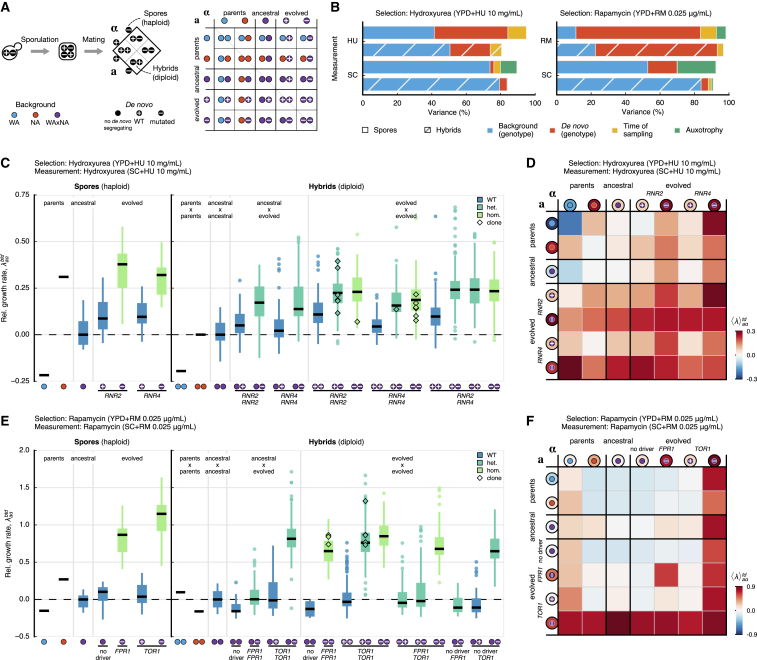
 ; NA parent,
; NA parent,  ; WAxNA segregant,
; WAxNA segregant,  ) and the genotype of de novo mutations (no de novo mutation,
) and the genotype of de novo mutations (no de novo mutation,  ; wild-type,
; wild-type,  ; mutated,
; mutated,  ). An extended version of the figure with all combinations and controls can be found in Figures S10 and S11, respectively.
). An extended version of the figure with all combinations and controls can be found in Figures S10 and S11, respectively.References
-
- Balaban N.Q., Merrin J., Chait R., Kowalik L., Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. - PubMed
-
- Burke M.K., Dunham J.P., Shahrestani P., Thornton K.R., Rose M.R., Long A.D. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature. 2010;467:587–590. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
