

Revealing the Determinants of Widespread Alternative Splicing Perturbation in Cancer
- PMID: 29045845
- PMCID: PMC5689467
- DOI: 10.1016/j.celrep.2017.09.071
Revealing the Determinants of Widespread Alternative Splicing Perturbation in Cancer
Abstract
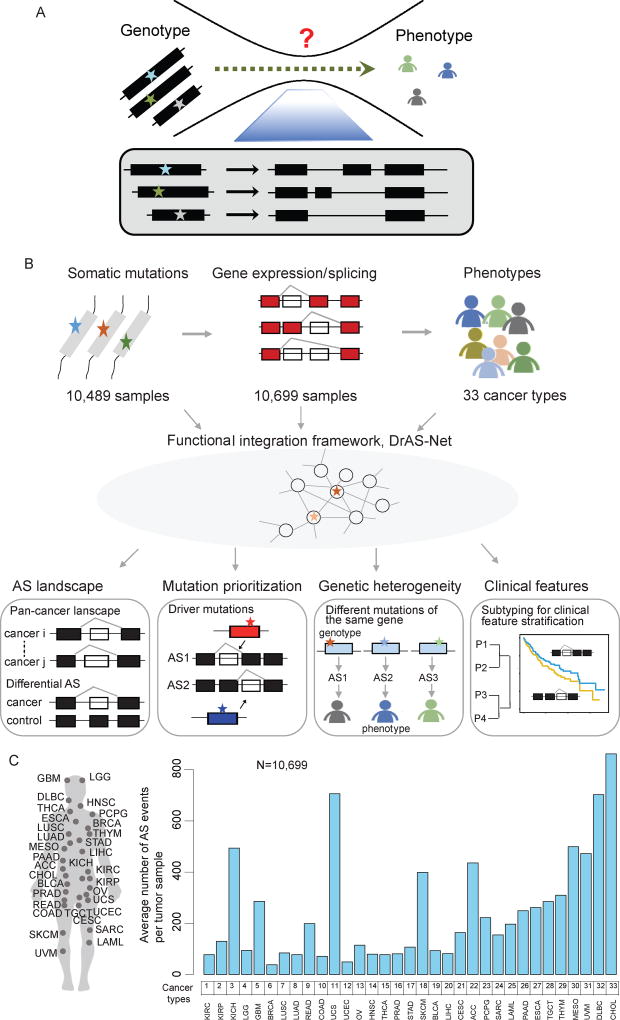
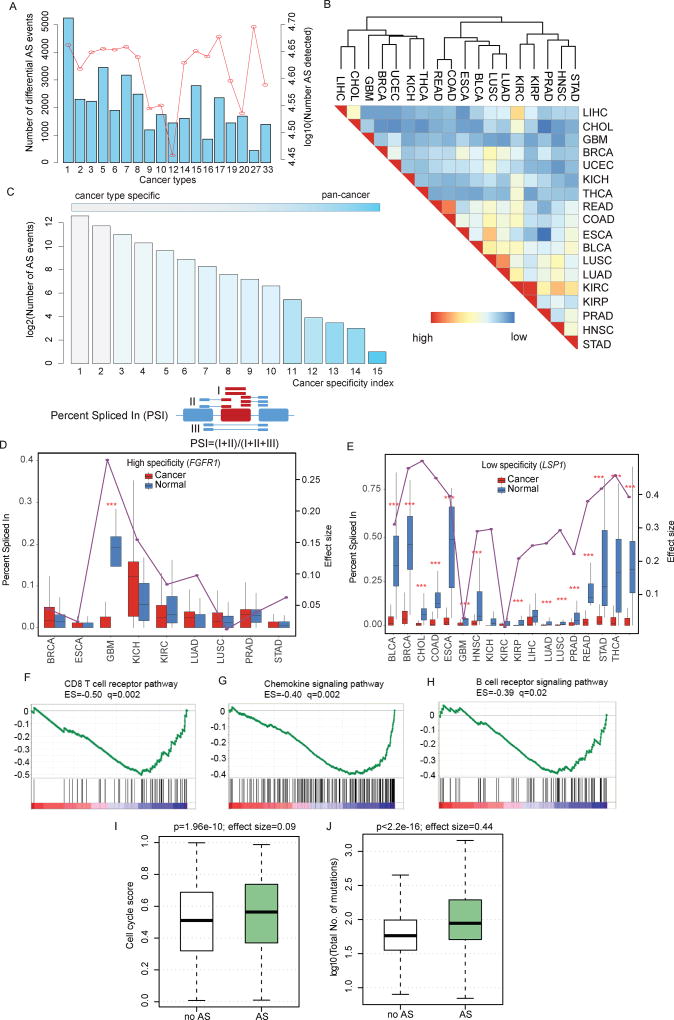
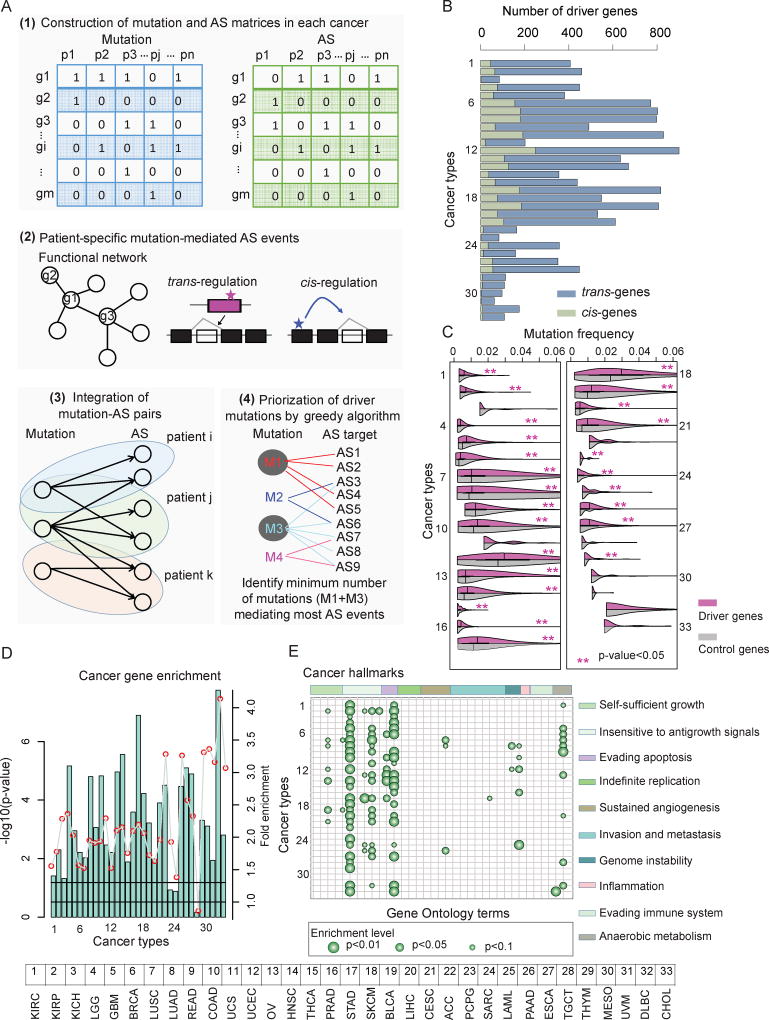
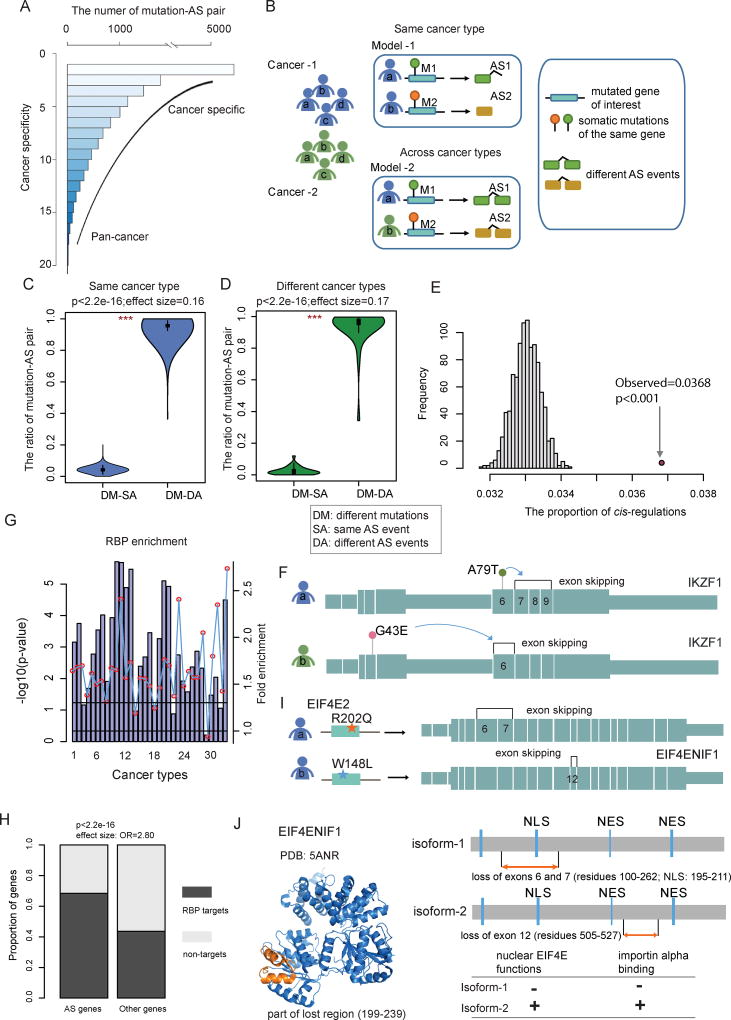
It is increasingly appreciated that alternative splicing plays a key role in generating functional specificity and diversity in cancer. However, the mechanisms by which cancer mutations perturb splicing remain unknown. Here, we developed a network-based strategy, DrAS-Net, to investigate more than 2.5 million variants across cancer types and link somatic mutations with cancer-specific splicing events. We identified more than 40,000 driver variant candidates and their 80,000 putative splicing targets deregulated in 33 cancer types and inferred their functional impact. Strikingly, tumors with splicing perturbations show reduced expression of immune system-related genes and increased expression of cell proliferation markers. Tumors harboring different mutations in the same gene often exhibit distinct splicing perturbations. Further stratification of 10,000 patients based on their mutation-splicing relationships identifies subtypes with distinct clinical features, including survival rates. Our work reveals how single-nucleotide changes can alter the repertoires of splicing isoforms, providing insights into oncogenic mechanisms for precision medicine.
Keywords: DrAS-Net; Network biology; alternative splicing; bioinformatics; cancer; computational biology; gene regulation; genotype-phenotype relationships; somatic mutations; systems biology.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
