

Impacts of Genome-Wide Analyses on Our Understanding of Human Herpesvirus Diversity and Evolution
- PMID: 29046445
- PMCID: PMC5730764
- DOI: 10.1128/JVI.00908-17
Impacts of Genome-Wide Analyses on Our Understanding of Human Herpesvirus Diversity and Evolution
Abstract
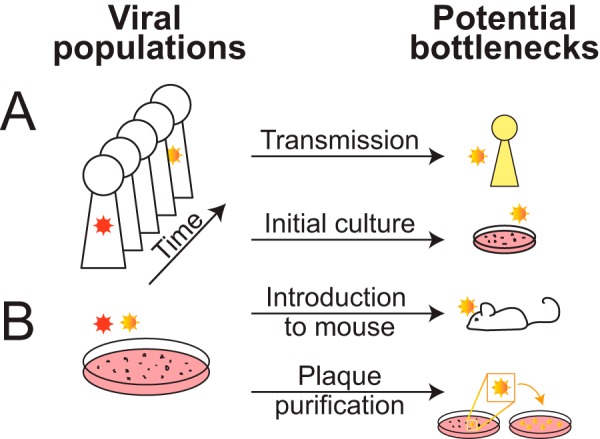
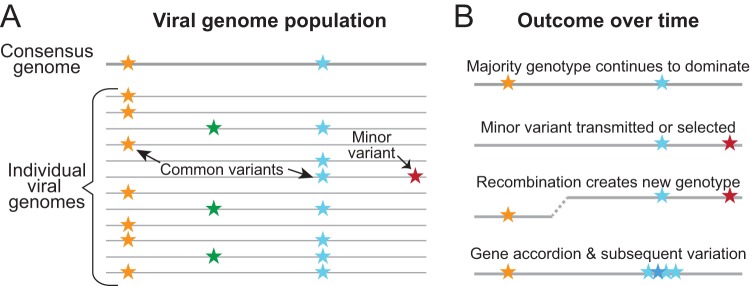
Until fairly recently, genome-wide evolutionary dynamics and within-host diversity were more commonly examined in the context of small viruses than in the context of large double-stranded DNA viruses such as herpesviruses. The high mutation rates and more compact genomes of RNA viruses have inspired the investigation of population dynamics for these species, and recent data now suggest that herpesviruses might also be considered candidates for population modeling. High-throughput sequencing (HTS) and bioinformatics have expanded our understanding of herpesviruses through genome-wide comparisons of sequence diversity, recombination, allele frequency, and selective pressures. Here we discuss recent data on the mechanisms that generate herpesvirus genomic diversity and underlie the evolution of these virus families. We focus on human herpesviruses, with key insights drawn from veterinary herpesviruses and other large DNA virus families. We consider the impacts of cell culture on herpesvirus genomes and how to accurately describe the viral populations under study. The need for a strong foundation of high-quality genomes is also discussed, since it underlies all secondary genomic analyses such as RNA sequencing (RNA-Seq), chromatin immunoprecipitation, and ribosome profiling. Areas where we foresee future progress, such as the linking of viral genetic differences to phenotypic or clinical outcomes, are highlighted as well.
Keywords: bioinformatics; diversity; evolution; genetic recombination; genomics; herpesvirus; minority variant; polymorphism; viral pathogenesis.
Copyright © 2017 Renner and Szpara.
Figures


References
-
- Jansen MAE, van den Heuvel D, Bouthoorn SH, Jaddoe VWV, Hooijkaas H, Raat H, Fraaij PLA, van Zelm MC, Moll HA. 2016. Determinants of ethnic differences in cytomegalovirus, Epstein-Barr virus, and herpes simplex virus type 1 seroprevalence in childhood. J Pediatr 170:126–134.e6. doi:10.1016/j.jpeds.2015.11.014. - DOI - PubMed
-
- Gantt S, Orem J, Krantz EM, Morrow RA, Selke S, Huang M-L, Schiffer JT, Jerome KR, Nakaganda A, Wald A, Casper C, Corey L. 24 February 2016. Prospective characterization of the risk factors for transmission and symptoms of primary human herpesvirus infections among Ugandan infants. J Infect Dis doi:10.1093/infdis/jiw076. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
