

NMNAT3 is protective against the effects of neonatal cerebral hypoxia-ischemia
- PMID: 29046881
- PMCID: PMC5634348
- DOI: 10.1002/acn3.450
NMNAT3 is protective against the effects of neonatal cerebral hypoxia-ischemia
Abstract
Objective: To determine whether the NAD+ biosynthetic protein, nicotinamide mononucleotide adenylyltransferase-3 (NMNAT3), is a neuroprotective inducible enzyme capable of decreasing cerebral injury after neonatal hypoxia-ischemia (H-I) and reducing glutamate receptor-mediated excitotoxic neurodegeneration of immature neurons.
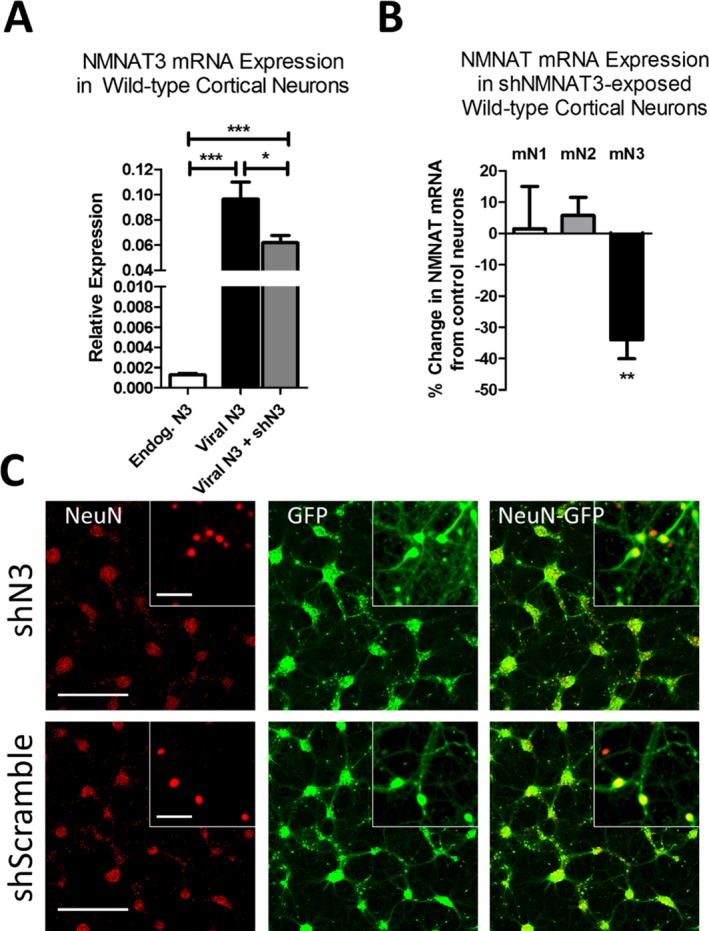
Methods: Using NMNAT3-overexpressing mice we investigated whether increases in brain NMNAT3 reduced cerebral tissue loss following H-I. We then employed biochemical methods from injured neonatal brains to examine the inducibility of NMNAT3 and the mechanism of NMNAT3-dependent neuroprotection. Using AAV8-mediated vectors for in vitro neuronal NMNAT3 knockdown, we then examine the endogenous role of this protein on immature neuronal survival prior and following NMDA receptor-mediated excitotoxicity.
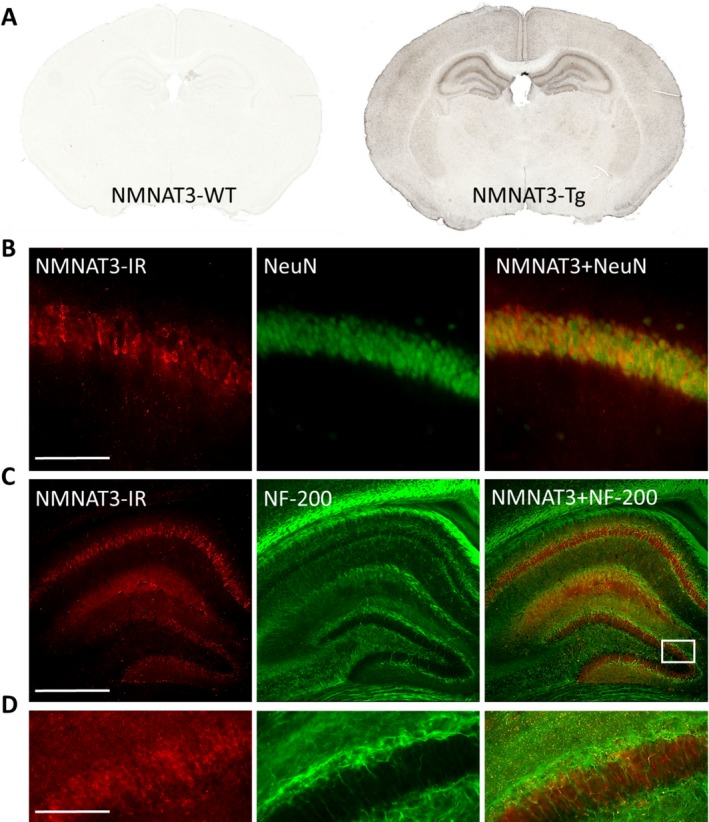
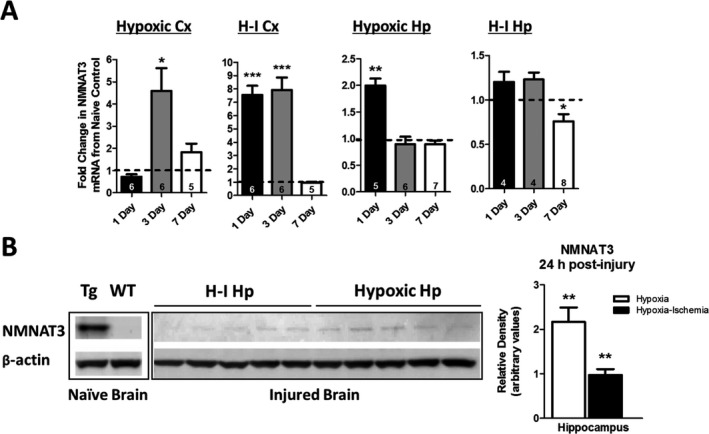
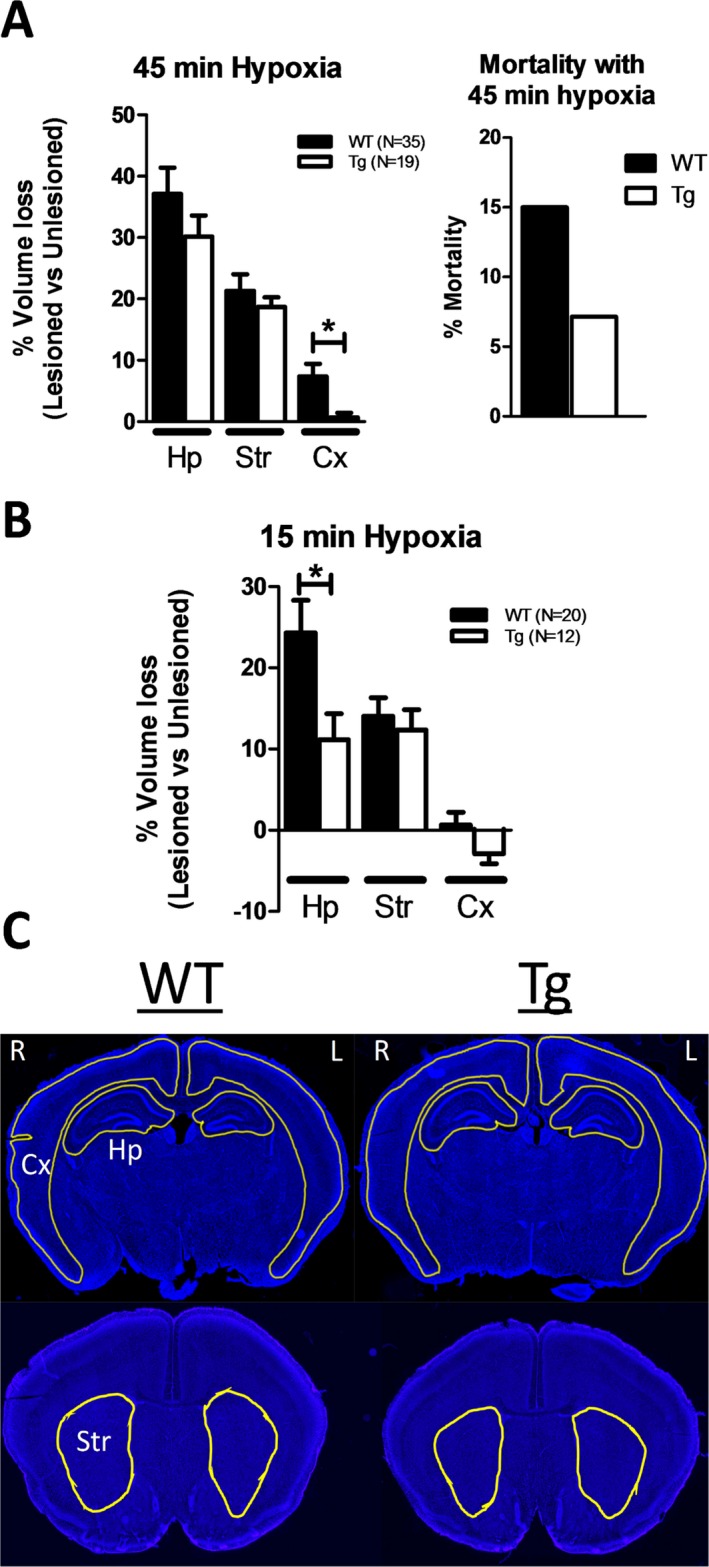
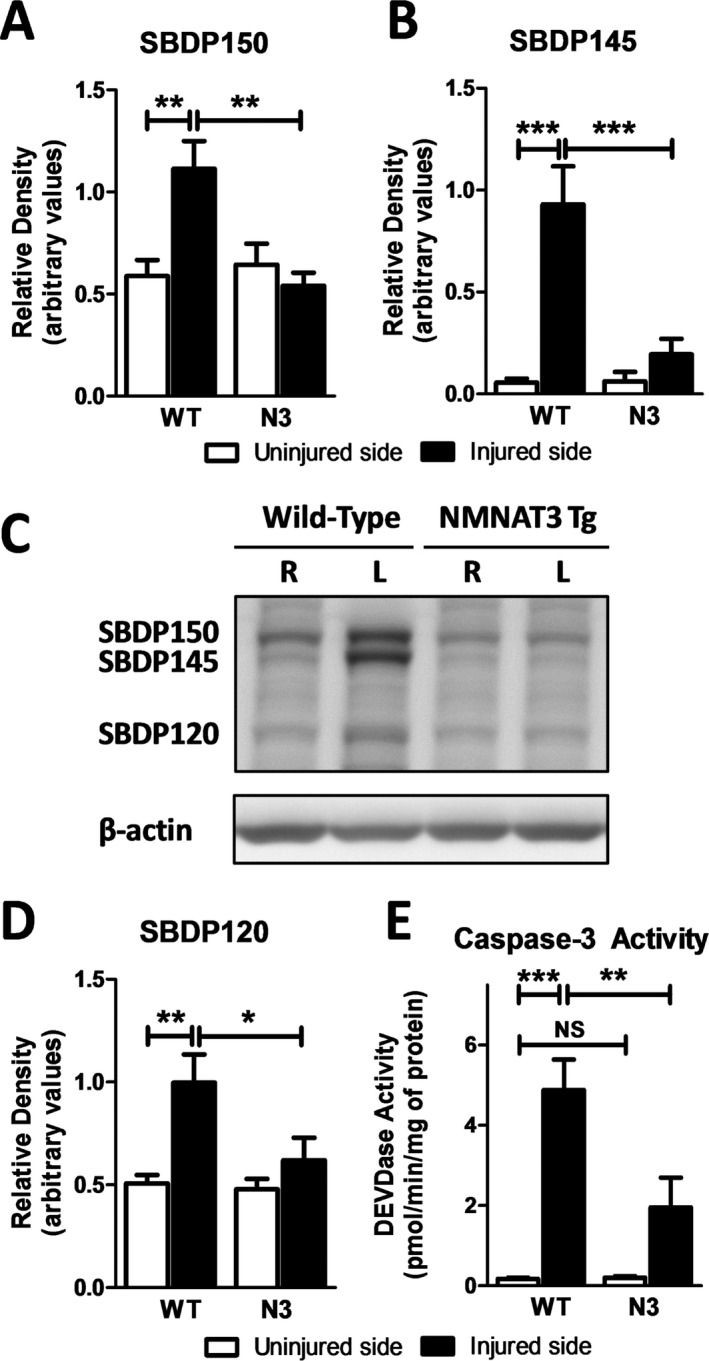
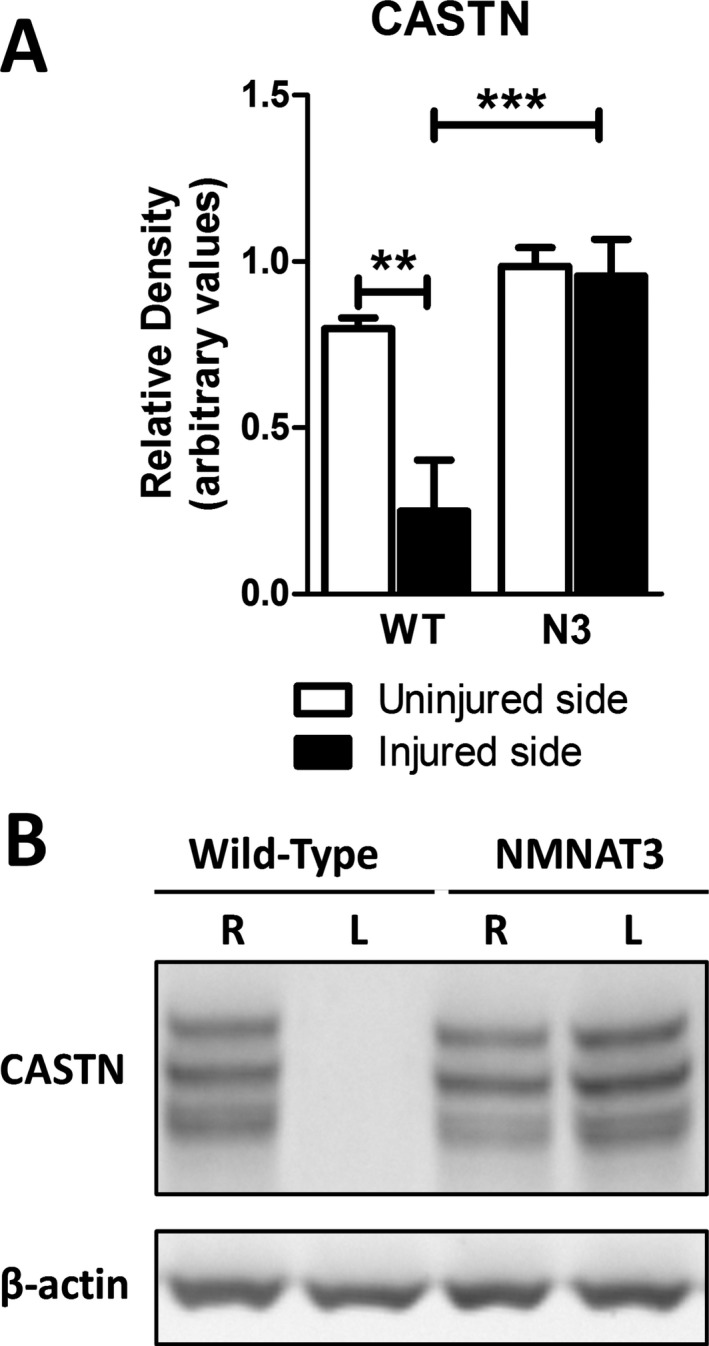
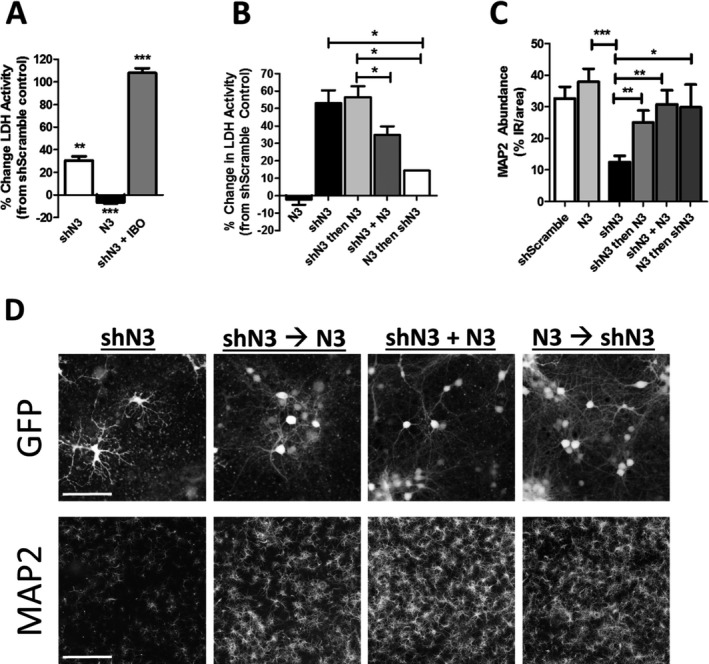
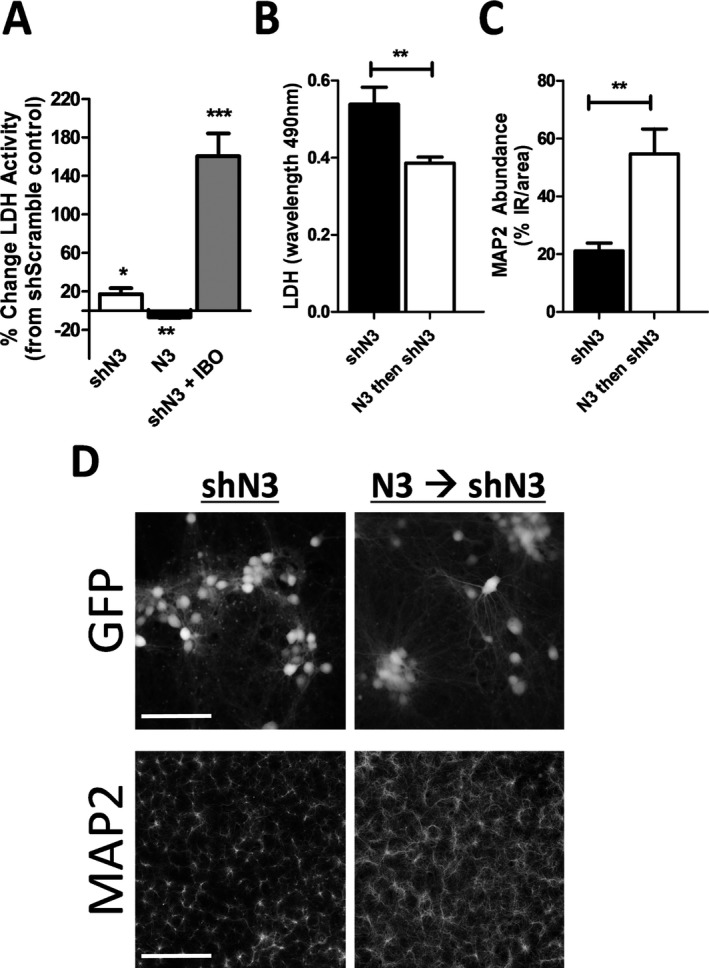
Results: NMNAT3 mRNA and protein levels increased after neonatal H-I. In addition, NMNAT3 overexpression decreased cortical and hippocampal tissue loss 7 days following injury. We further show that the NMNAT3 neuroprotective mechanism involves a decrease in calpastatin degradation, and a decrease in caspase-3 activity and calpain-mediated cleavage. Conversely, NMNAT3 knockdown of cortical and hippocampal neurons in vitro caused neuronal degeneration and increased excitotoxic cell death. The neurodegenerative effects of NMNAT3 knockdown were counteracted by exogenous upregulation of NMNAT3.
Conclusions: Our observations provide new insights into the neuroprotective mechanisms of NMNATs in the injured developing brain, adding NMNAT3 as an important neuroprotective enzyme in neonatal H-I via inhibition of apoptotic and necrotic neurodegeneration. Interestingly, we find that endogenous NMNAT3 is an inducible protein important for maintaining the survival of immature neurons. Future studies aimed at uncovering the mechanisms of NMNAT3 upregulation and neuroprotection may offer new therapies against the effects of hypoxic-ischemic encephalopathy.
Figures
Similar articles
-
Nicotinamide mononucleotide adenylyl transferase 1 protects against acute neurodegeneration in developing CNS by inhibiting excitotoxic-necrotic cell death.Proc Natl Acad Sci U S A. 2011 Nov 22;108(47):19054-9. doi: 10.1073/pnas.1107325108. Epub 2011 Nov 4. Proc Natl Acad Sci U S A. 2011. PMID: 22058226 Free PMC article.
-
Pretreatment with Human Chorionic Gonadotropin Protects the Neonatal Brain against the Effects of Hypoxic-Ischemic Injury.Front Pediatr. 2017 Nov 3;5:232. doi: 10.3389/fped.2017.00232. eCollection 2017. Front Pediatr. 2017. PMID: 29164084 Free PMC article.
-
Subcellular NAMPT-mediated NAD+ salvage pathways and their roles in bioenergetics and neuronal protection after ischemic injury.J Neurochem. 2019 Dec;151(6):732-748. doi: 10.1111/jnc.14878. Epub 2019 Oct 16. J Neurochem. 2019. PMID: 31553812 Free PMC article.
-
Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis.Brain Res Bull. 1998 Jul 1;46(4):281-309. doi: 10.1016/s0361-9230(98)00024-0. Brain Res Bull. 1998. PMID: 9671259 Review.
-
Molecular mediators of hypoxic-ischemic injury and implications for epilepsy in the developing brain.Epilepsy Behav. 2005 Sep;7(2):204-13. doi: 10.1016/j.yebeh.2005.05.015. Epilepsy Behav. 2005. PMID: 16054439 Review.
Cited by
-
Human Nmnat1 Promotes Autophagic Clearance of Amyloid Plaques in a Drosophila Model of Alzheimer's Disease.Front Aging Neurosci. 2022 Mar 24;14:852972. doi: 10.3389/fnagi.2022.852972. eCollection 2022. Front Aging Neurosci. 2022. PMID: 35401143 Free PMC article.
-
Molecular mechanisms of excitotoxicity and their relevance to the pathogenesis of neurodegenerative diseases-an update.Acta Pharmacol Sin. 2025 May 19. doi: 10.1038/s41401-025-01576-w. Online ahead of print. Acta Pharmacol Sin. 2025. PMID: 40389567 Review.
-
Functional neuropathology of neonatal hypoxia-ischemia by single-mouse longitudinal electroencephalography.Epilepsia. 2022 Dec;63(12):3037-3050. doi: 10.1111/epi.17403. Epub 2022 Oct 12. Epilepsia. 2022. PMID: 36054439 Free PMC article.
-
Positive and negative conditioning in the neonatal brain.Cond Med. 2018 Oct;1(6):279-293. Cond Med. 2018. PMID: 31214666 Free PMC article.
-
Pathobiochemistry of Aging and Neurodegeneration: Deregulation of NAD+ Metabolism in Brain Cells.Biomolecules. 2024 Dec 6;14(12):1556. doi: 10.3390/biom14121556. Biomolecules. 2024. PMID: 39766263 Free PMC article. Review.
References
-
- Ferriero DM. Neonatal brain injury. N Engl J Med 2004;351:1985–1995. - PubMed
-
- Vexler ZS, Ferriero DM. Molecular and biochemical mechanisms of perinatal brain injury. Semin Neonatol 2001;6:99–108. - PubMed
-
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 2005;280:36334–36341. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
