

Endothelial Cell Reactive Oxygen Species and Ca2+ Signaling in Pulmonary Hypertension
- PMID: 29047094
- PMCID: PMC7398047
- DOI: 10.1007/978-3-319-63245-2_18
Endothelial Cell Reactive Oxygen Species and Ca2+ Signaling in Pulmonary Hypertension
Abstract
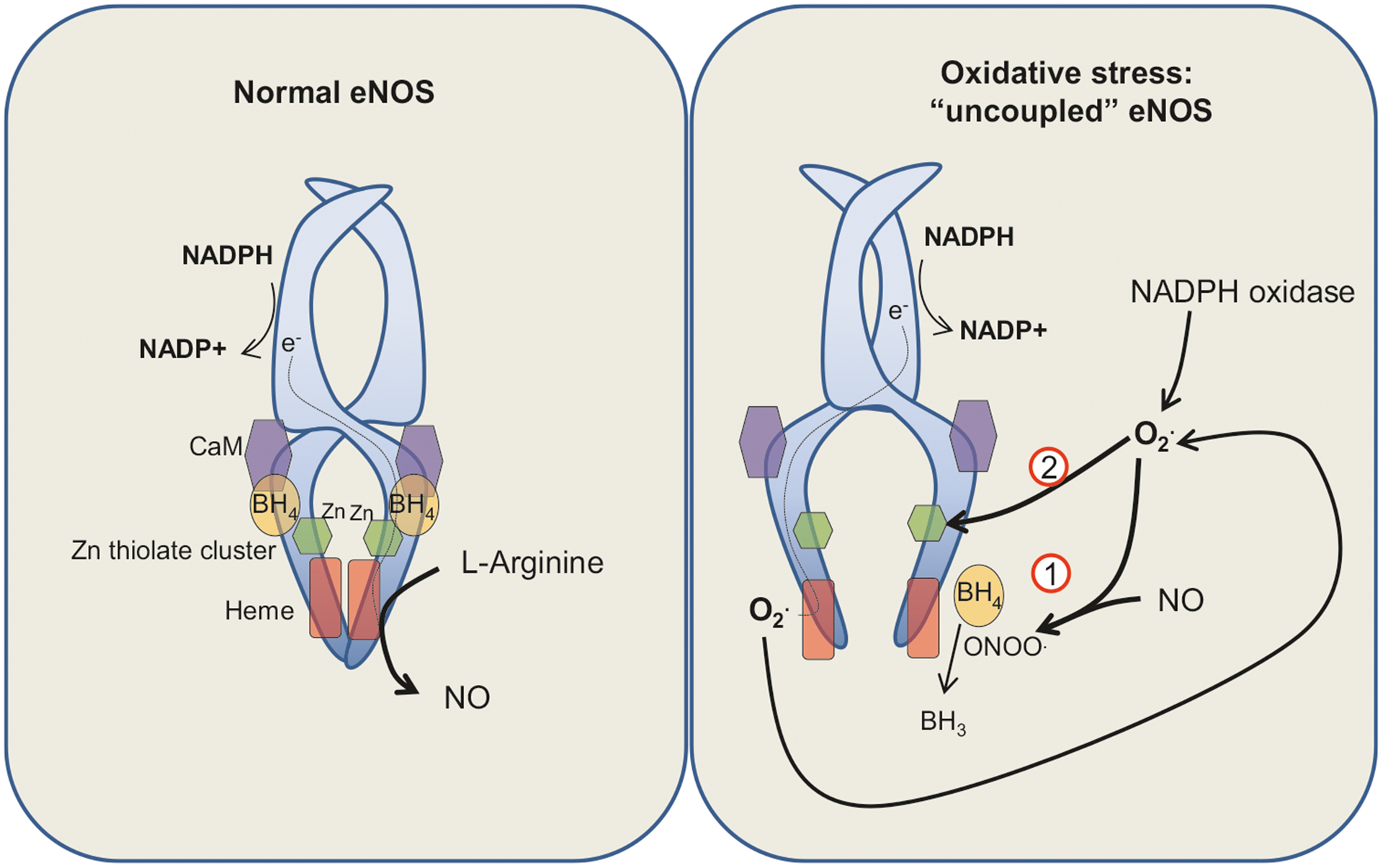
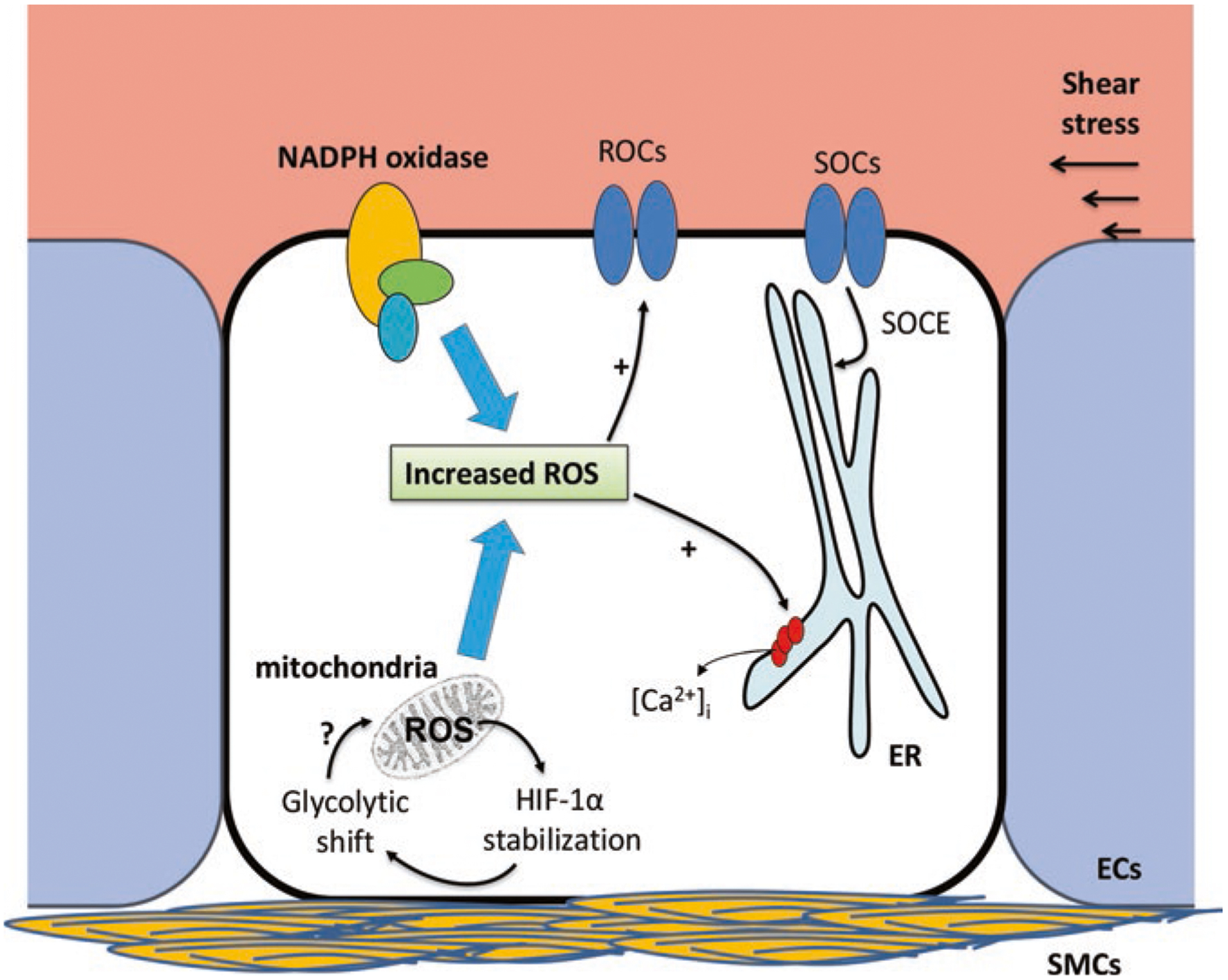
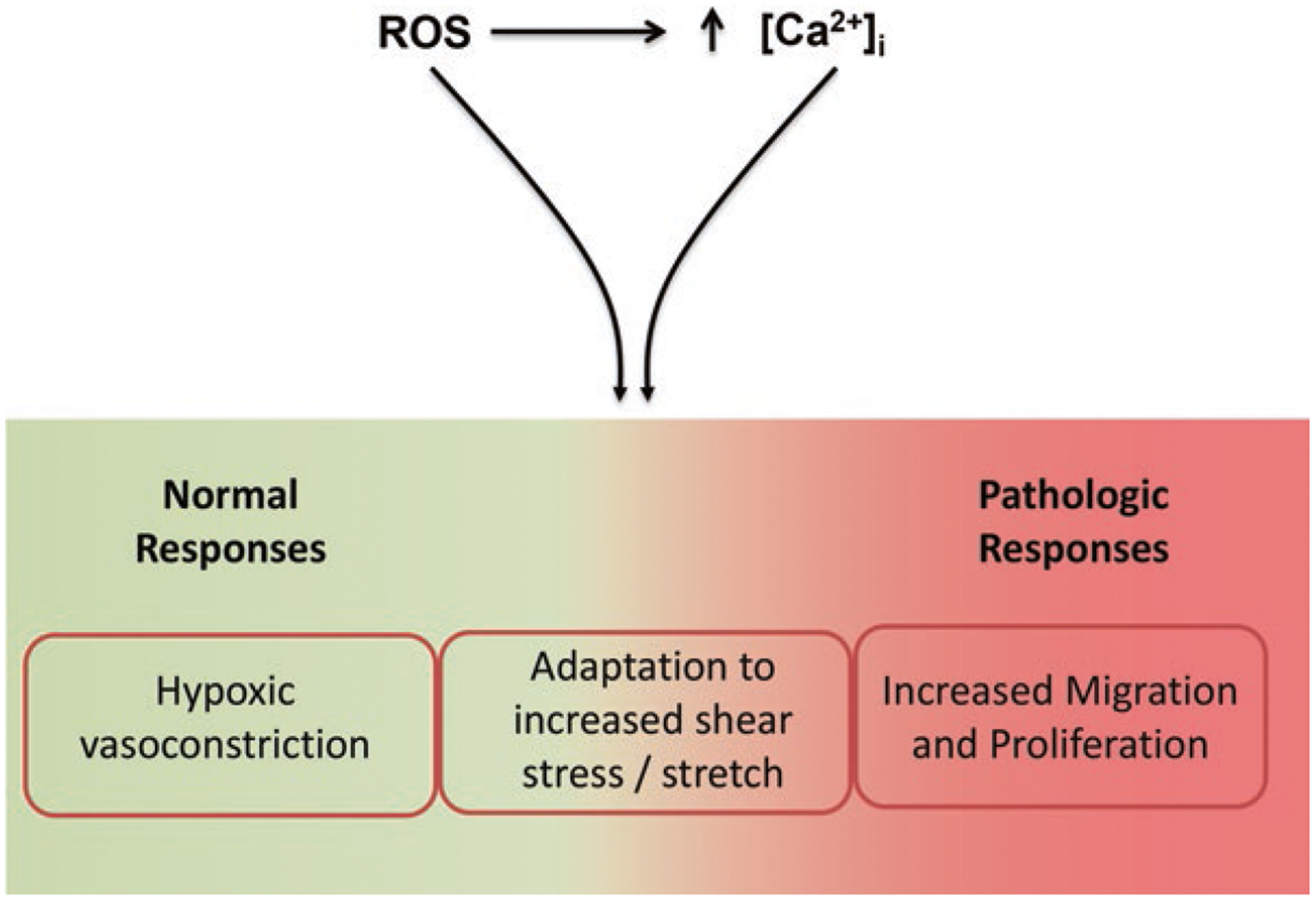
Pulmonary hypertension (PH) refers to a disorder characterized by elevated pulmonary arterial pressure, leading to right ventricular overload and eventually right ventricular failure, which results in high morbidity and mortality. PH is associated with heterogeneous etiologies and distinct molecular mechanisms, including abnormal migration and proliferation of endothelial and smooth muscle cells. Although the exact details are not fully elucidated, reactive oxygen species (ROS) have been shown to play a key role in promoting abnormal function in pulmonary arterial smooth muscle and endothelial cells in PH. In endothelial cells, ROS can be generated from sources such as NADPH oxidase and mitochondria, which in turn can serve as signaling molecules in a wide variety of processes including posttranslational modification of proteins involved in Ca2+ homeostasis. In this chapter, we discuss the role of ROS in promoting abnormal vasoreactivity and endothelial migration and proliferation in various models of PH. Furthermore, we draw particular attention to the role of ROS-induced increases in intracellular Ca2+ concentration in the pathobiology of PH.
Keywords: Calcium homeostasis; Endothelial cell; Hypoxia; Mitochondria; NADPH oxidase; Pulmonary hypertension; Reactive oxygen species.
Figures
Similar articles
-
Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension.Free Radic Biol Med. 2015 Oct;87:36-47. doi: 10.1016/j.freeradbiomed.2015.05.042. Epub 2015 Jun 12. Free Radic Biol Med. 2015. PMID: 26073127 Free PMC article.
-
Cross Talk Between Mitochondrial Reactive Oxygen Species and Sarcoplasmic Reticulum Calcium in Pulmonary Arterial Smooth Muscle Cells.Adv Exp Med Biol. 2017;967:289-298. doi: 10.1007/978-3-319-63245-2_17. Adv Exp Med Biol. 2017. PMID: 29047093 Review.
-
Endothelial Nox1 oxidase assembly in human pulmonary arterial hypertension; driver of Gremlin1-mediated proliferation.Clin Sci (Lond). 2017 Jul 16;131(15):2019-2035. doi: 10.1042/CS20160812. Print 2017 Aug 1. Clin Sci (Lond). 2017. PMID: 28522681 Free PMC article.
-
Role of Transcription Factors in Pulmonary Artery Smooth Muscle Cells: An Important Link to Hypoxic Pulmonary Hypertension.Adv Exp Med Biol. 2017;967:13-32. doi: 10.1007/978-3-319-63245-2_2. Adv Exp Med Biol. 2017. PMID: 29047078
-
Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction.Eur Respir J. 2016 Jan;47(1):288-303. doi: 10.1183/13993003.00945-2015. Epub 2015 Oct 22. Eur Respir J. 2016. PMID: 26493804 Review.
Cited by
-
Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime?Int J Mol Sci. 2021 Sep 10;22(18):9821. doi: 10.3390/ijms22189821. Int J Mol Sci. 2021. PMID: 34575985 Free PMC article. Review.
-
Heart‑lung crosstalk in pulmonary arterial hypertension following myocardial infarction (Review).Int J Mol Med. 2020 Sep;46(3):913-924. doi: 10.3892/ijmm.2020.4650. Epub 2020 Jun 18. Int J Mol Med. 2020. PMID: 32582962 Free PMC article. Review.
-
Cellular and Molecular Processes in Pulmonary Hypertension.Adv Exp Med Biol. 2021;1304:21-38. doi: 10.1007/978-3-030-68748-9_2. Adv Exp Med Biol. 2021. PMID: 34019261
-
Effects of FW2 Nanoparticles Toxicity in a New In Vitro Pulmonary Vascular Cells Model Mimicking Endothelial Dysfunction.Cardiovasc Toxicol. 2022 Jan;22(1):14-28. doi: 10.1007/s12012-021-09679-6. Epub 2021 Sep 15. Cardiovasc Toxicol. 2022. PMID: 34524626
-
Transgenerational inheritance of promoter methylation changes in extrauterine growth restriction-induced pulmonary arterial pressure disorders.Ann Transl Med. 2021 Oct;9(20):1551. doi: 10.21037/atm-21-4715. Ann Transl Med. 2021. PMID: 34790757 Free PMC article.
References
-
- Touyz RM (2005). Reactive oxygen species as mediators of calcium signaling by angiotensin II: Implications in vascular physiology and patho-physiology. Antioxidants & Redox Signaling, 7, 1302–1314. - PubMed
-
- Gebb S, & Stevens T (2004). On lung endothelial cell heterogeneity. Microvascular Research, 68, 1–12. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
