

Indexcov: fast coverage quality control for whole-genome sequencing
- PMID: 29048539
- PMCID: PMC5737511
- DOI: 10.1093/gigascience/gix090
Indexcov: fast coverage quality control for whole-genome sequencing
Abstract
The BAM and CRAM formats provide a supplementary linear index that facilitates rapid access to sequence alignments in arbitrary genomic regions. Comparing consecutive entries in a BAM or CRAM index allows one to infer the number of alignment records per genomic region for use as an effective proxy of sequence depth in each genomic region. Based on these properties, we have developed indexcov, an efficient estimator of whole-genome sequencing coverage to rapidly identify samples with aberrant coverage profiles, reveal large-scale chromosomal anomalies, recognize potential batch effects, and infer the sex of a sample. Indexcov is available at https://github.com/brentp/goleft under the MIT license.
© The Authors 2017. Published by Oxford University Press.
Figures
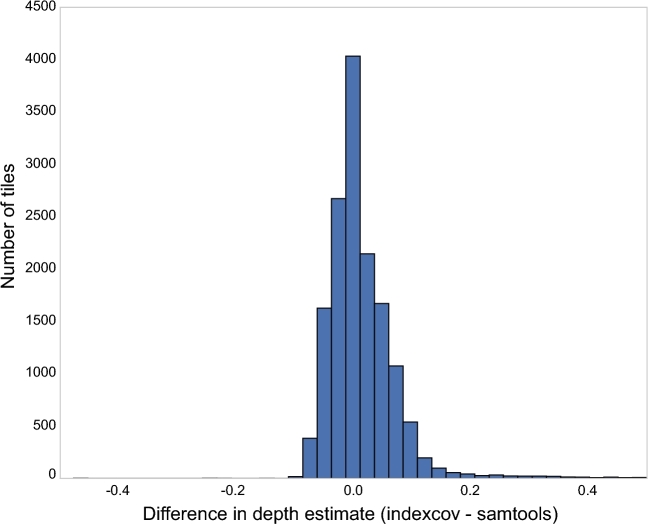
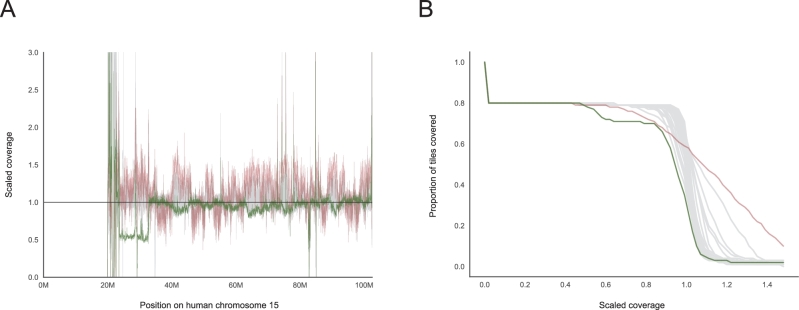

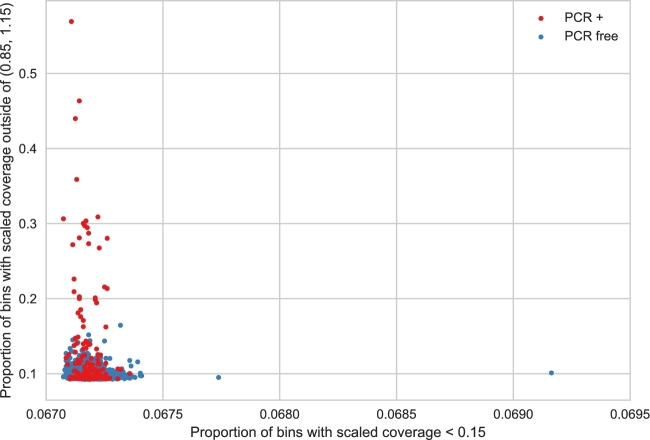
References
-
- Samtools https://samtools.github.io/hts-specs/CRAMv3.pdf. Accessed 27 May 2017.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
