

A method to reduce ancestry related germline false positives in tumor only somatic variant calling
- PMID: 29052513
- PMCID: PMC5649057
- DOI: 10.1186/s12920-017-0296-8
A method to reduce ancestry related germline false positives in tumor only somatic variant calling
Abstract
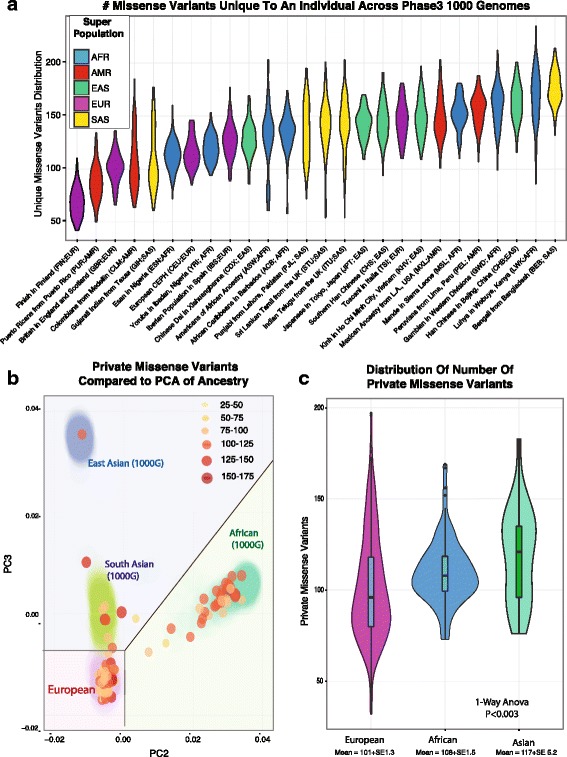
Background: Significant clinical and research applications are driving large scale adoption of individualized tumor sequencing in cancer in order to identify tumors-specific mutations. When a matched germline sample is available, somatic mutations may be identified using comparative callers. However, matched germline samples are frequently not available such as with archival tissues, which makes it difficult to distinguish somatic from germline variants. While population databases may be used to filter out known germline variants, recent studies have shown private germline variants result in an inflated false positive rate in unmatched tumor samples, and the number germline false positives in an individual may be related to ancestry.
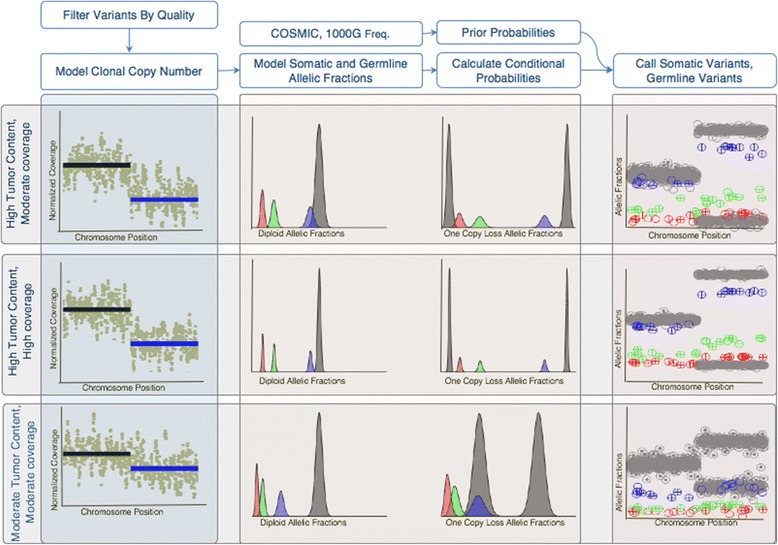
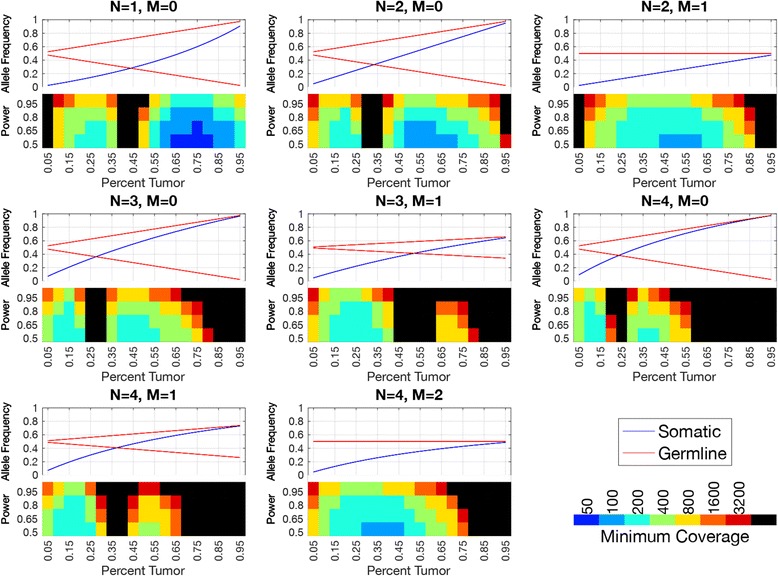
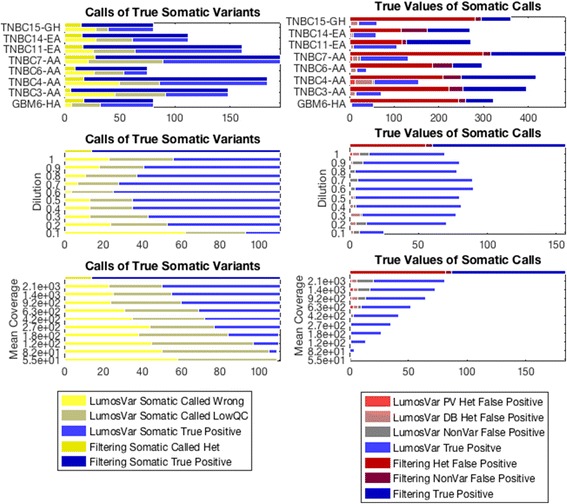
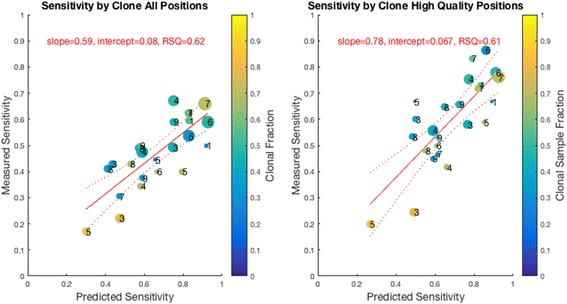
Methods: First, we examined the relationship between the germline false positives and ancestry. Then we developed and implemented a tumor only caller (LumosVar) that leverages differences in allelic frequency between somatic and germline variants in impure tumors. We used simulated data to systematically examine how copy number alterations, tumor purity, and sequencing depth should affect the sensitivity of our caller. Finally, we evaluated the caller on real data.
Results: We find the germline false-positive rate is significantly higher for individuals of non-European Ancestry largely due to the limited diversity in public polymorphism databases and due to population-specific characteristics such as admixture or recent expansions. Our Bayesian tumor only caller (LumosVar) is able to greatly reduce false positives from private germline variants, and our sensitivity is similar to predictions based on simulated data.
Conclusions: Taken together, our results suggest that studies of individuals of non-European ancestry would most benefit from our approach. However, high sensitivity requires sufficiently impure tumors and adequate sequencing depth. Even in impure tumors, there are copy number alterations that result in germline and somatic variants having similar allele frequencies, limiting the sensitivity of the approach. We believe our approach could greatly improve the analysis of archival samples in a research setting where the normal is not available.
Keywords: Cancer; Copy number alterations; Germline variant; Next generation sequencing; Precision medicine; Somatic mutation; Tumor purity.
Conflict of interest statement
Ethics approval and consent to participate
Only already existing de-identified data and biospecimens (both whole-blood and “fresh-frozen” tumor) previously collected under IRB approved studies (WIRB #20100721; WIRB #20141201; and WIRB #20031485) were used for this research.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Smith KS, Yadav VK, Pei S, Pollyea DA, Jordan CT, De S. SomVarIUS: somatic variant identification from unpaired tissue samples. Bioinformatics. 2015;2015:btv685. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
