

Neuro-ophthalmic side effects of molecularly targeted cancer drugs
- PMID: 29052609
- PMCID: PMC5811730
- DOI: 10.1038/eye.2017.222
Neuro-ophthalmic side effects of molecularly targeted cancer drugs
Abstract
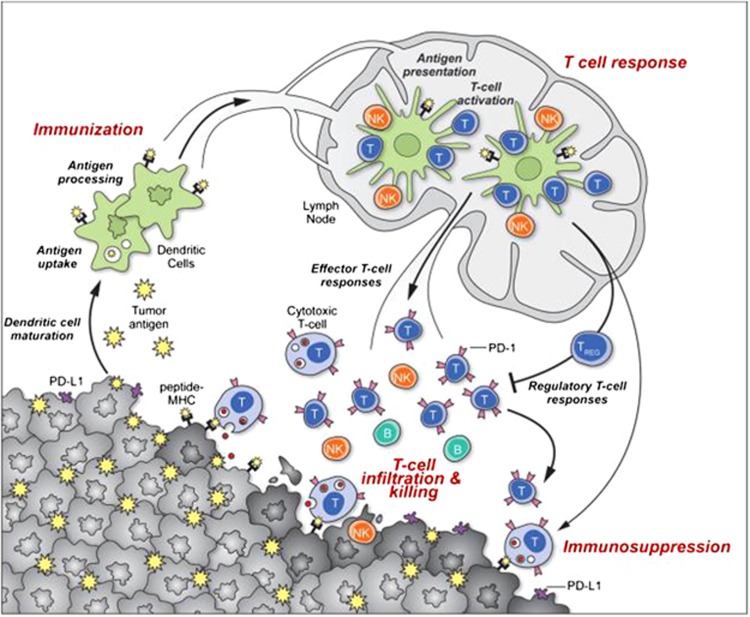
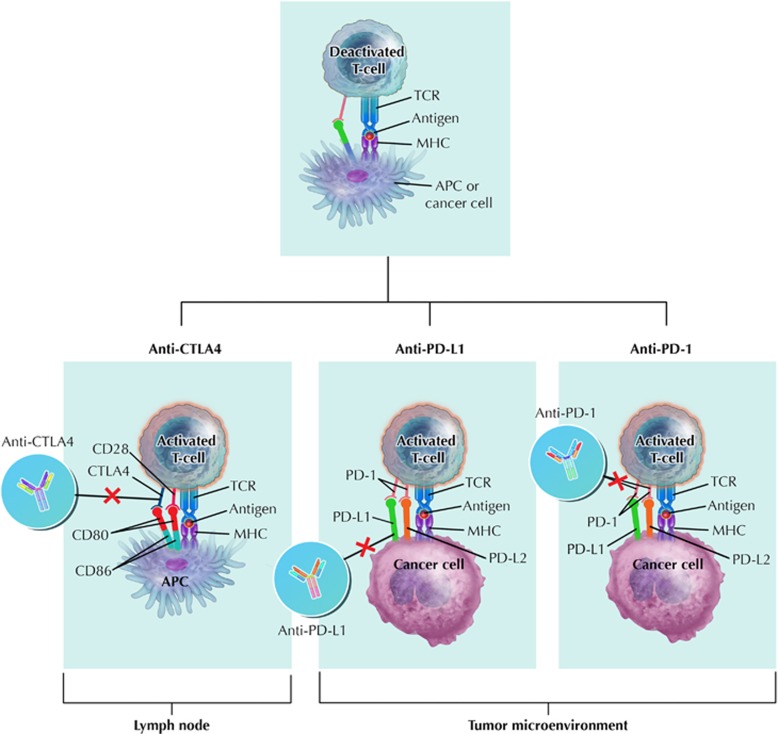
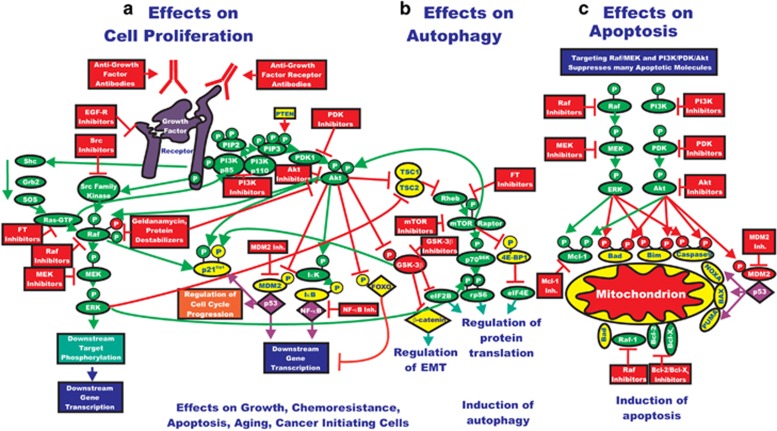
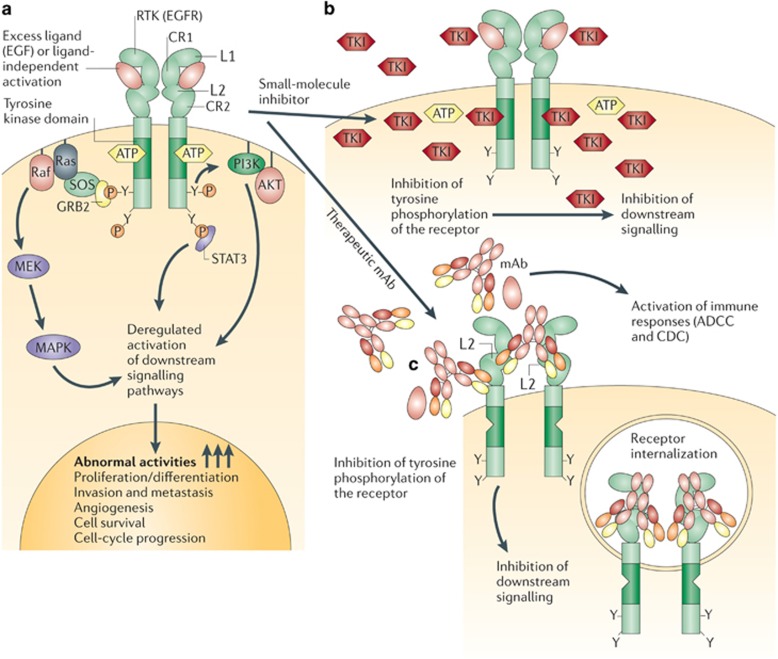
The past two decades has been an amazing time in the advancement of cancer treatment. Molecularly targeted therapy is a concept in which specific cellular molecules (overexpressed, mutationally activated, or selectively expressed proteins) are manipulated in an advantageous manner to decrease the transformation, proliferation, and/or survival of cancer cells. In addition, increased knowledge of the role of the immune system in carcinogenesis has led to the development of immune checkpoint inhibitors to restore and enhance cellular-mediated antitumor immunity. The United States Food and Drug Administration approval of the chimeric monoclonal antibody (mAb) rituximab in 1997 for the treatment of B cell non-Hodgkin lymphoma ushered in a new era of targeted therapy for cancer. A year later, trastuzumab, a humanized mAb, was approved for patients with breast cancer. In 2001, imatinib was the first small-molecule kinase inhibitor approved. The approval of ipilimumab-the first in class immune checkpoint inhibitor-in 2011 serves as a landmark period of time in the resurgence of immunotherapy for cancer. Despite the notion that increased tumor specificity results in decreased complications, toxicity remains a major hurdle in the development and implementation of many of the targeted anticancer drugs. This article will provide an overview of the current cellular and immunological understanding of cancer pathogenesis-the foundation upon which molecularly targeted therapies were developed-and a description of the ocular and neuro-ophthalmic toxicity profile of mAbs, immune checkpoint inhibitors, and small-molecule kinase inhibitors.
Conflict of interest statement
MTB: Novartis pharmaceuticals and Receptos.
AKSS: Bristol-Meyers Squibb, Celldex, Genentech, Merck, Immunocore, Reata.
Figures
References
-
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017; 67(1): 7–30. - PubMed
-
- Gerber DE. Targeted therapies: a new generation of cancer treatments. Am Fam Physician 2008; 77(3): 311–319. - PubMed
-
- Aggarwal S. Targeted cancer therapies. Nat Rev Drug Discov 2010; 9(6): 427–428. - PubMed
-
- Kocher R, Roberts B. The calculus of cures. N Engl J Med 2014; 370(16): 1473–1475. - PubMed
-
- Scharf O, Colevas AD. Adverse event reporting in publications compared with sponsor database for cancer clinical trials. J Clin Oncol 2006; 24(24): 3933–3938. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
