

Multiscale 3D Genome Rewiring during Mouse Neural Development
- PMID: 29053968
- PMCID: PMC5651218
- DOI: 10.1016/j.cell.2017.09.043
Multiscale 3D Genome Rewiring during Mouse Neural Development
Abstract
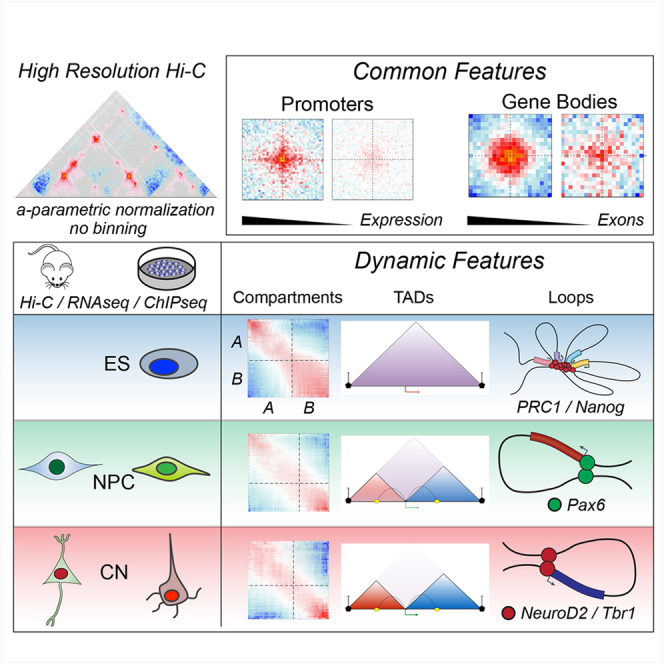
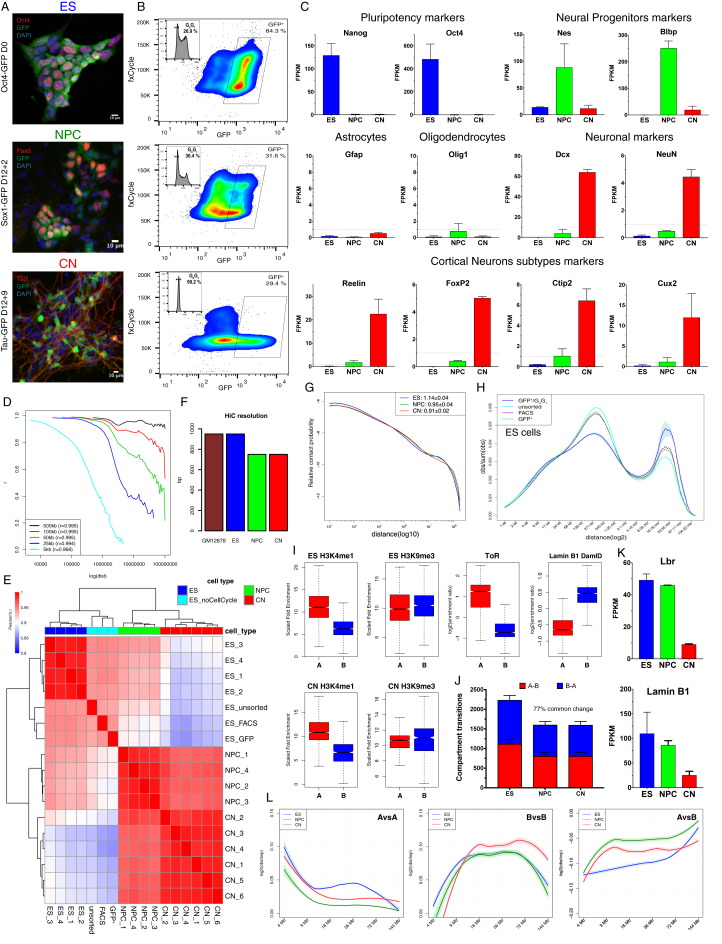
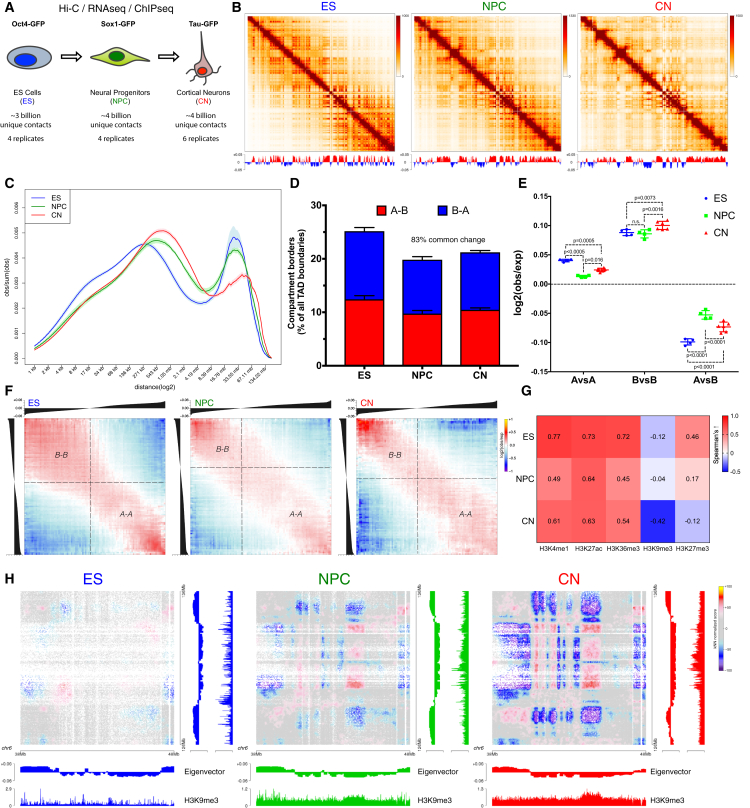
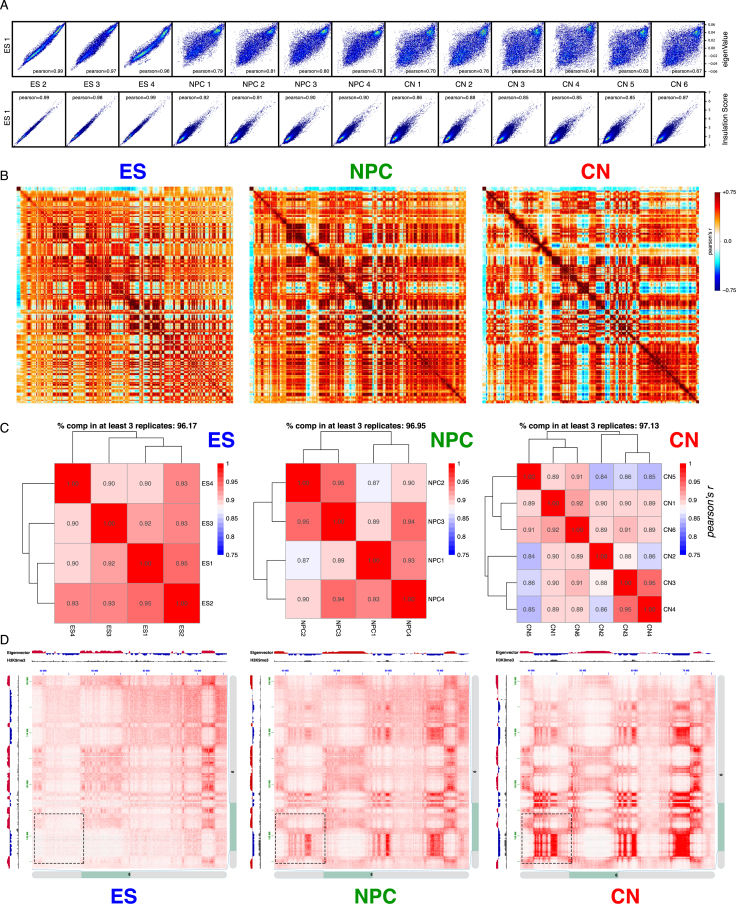
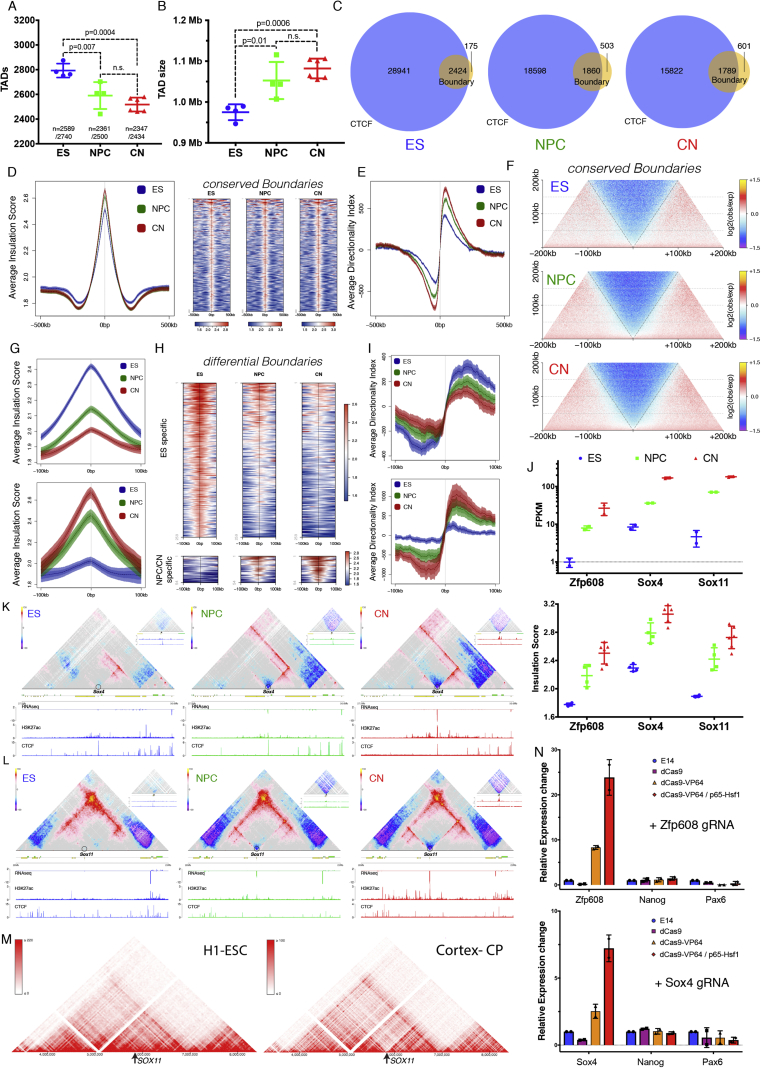
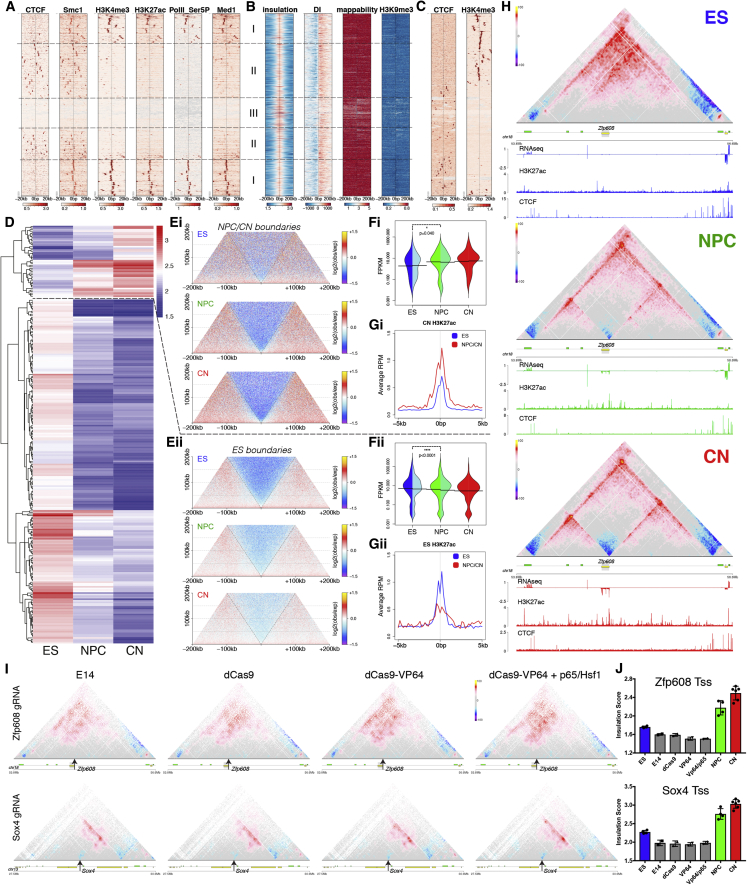
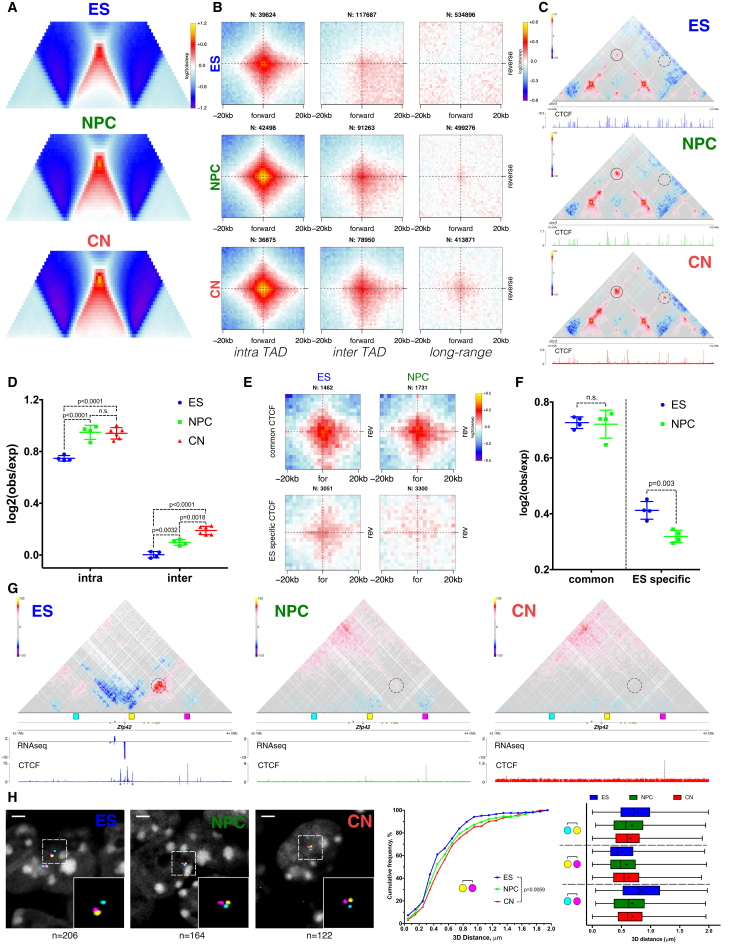
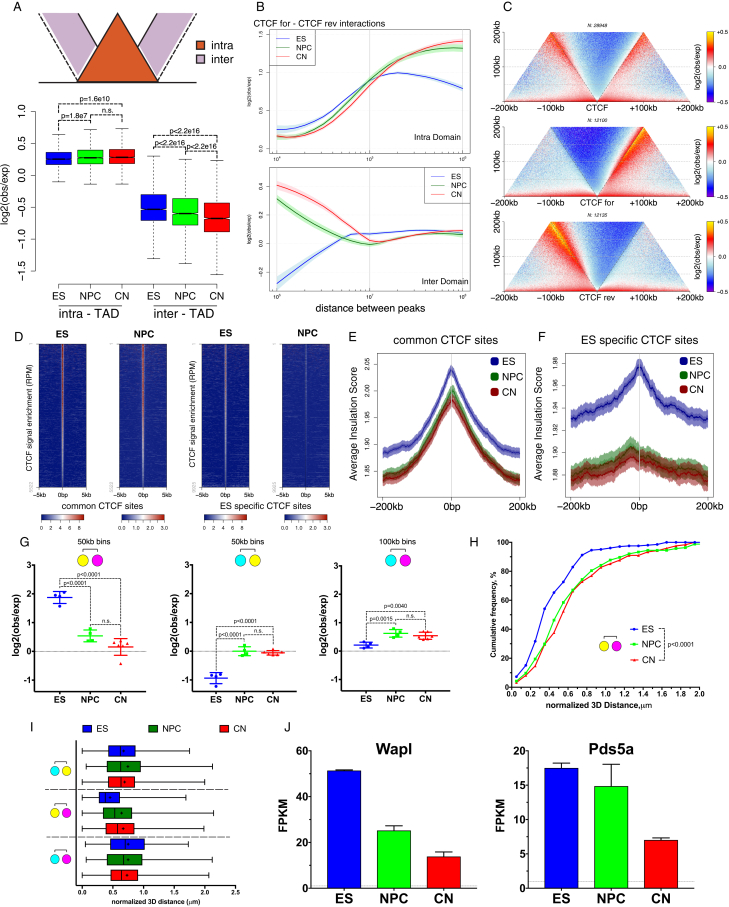
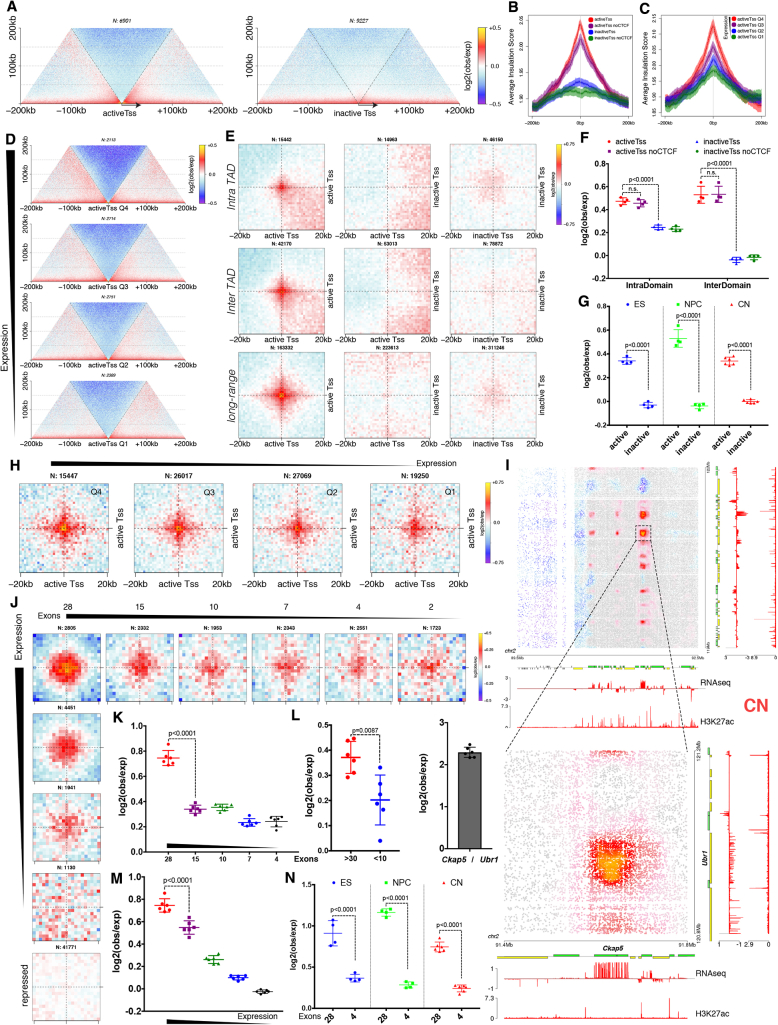
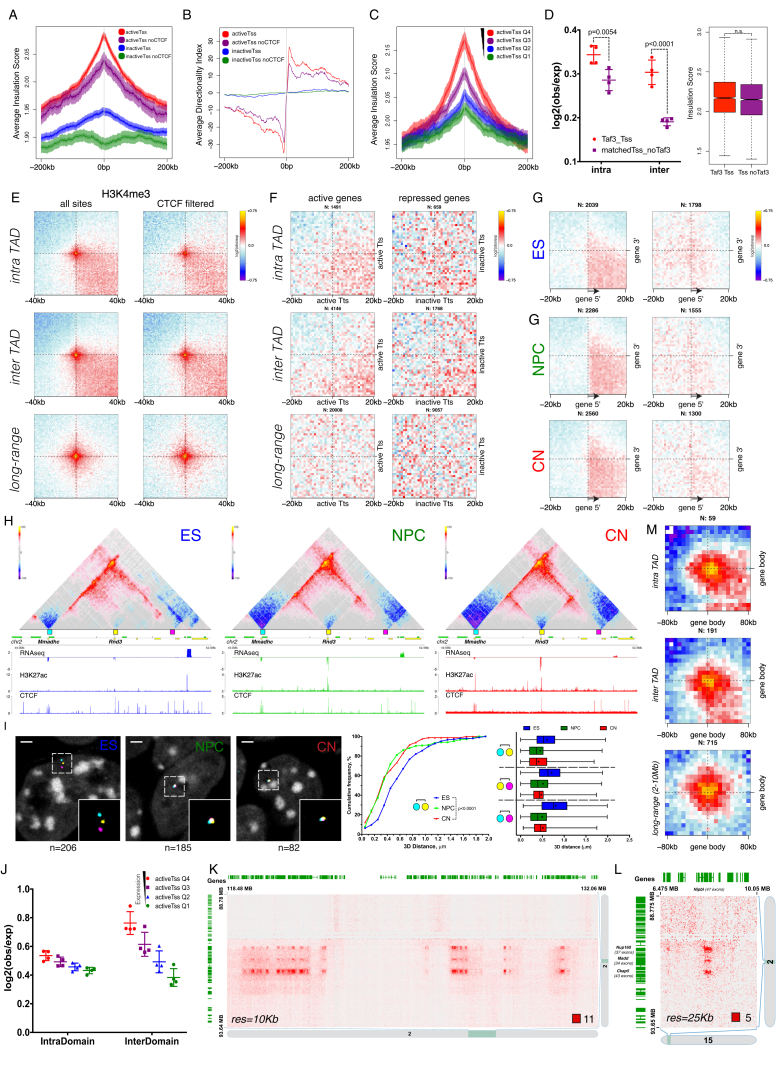
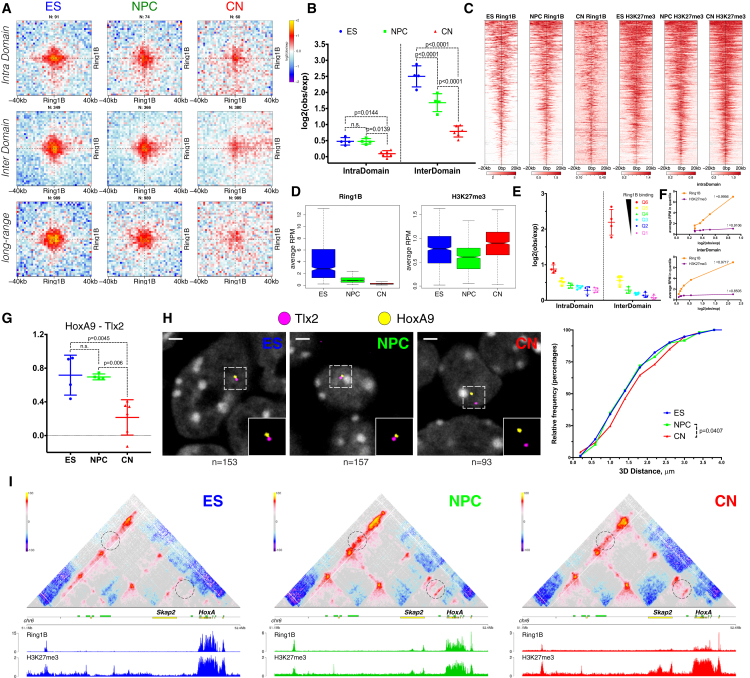
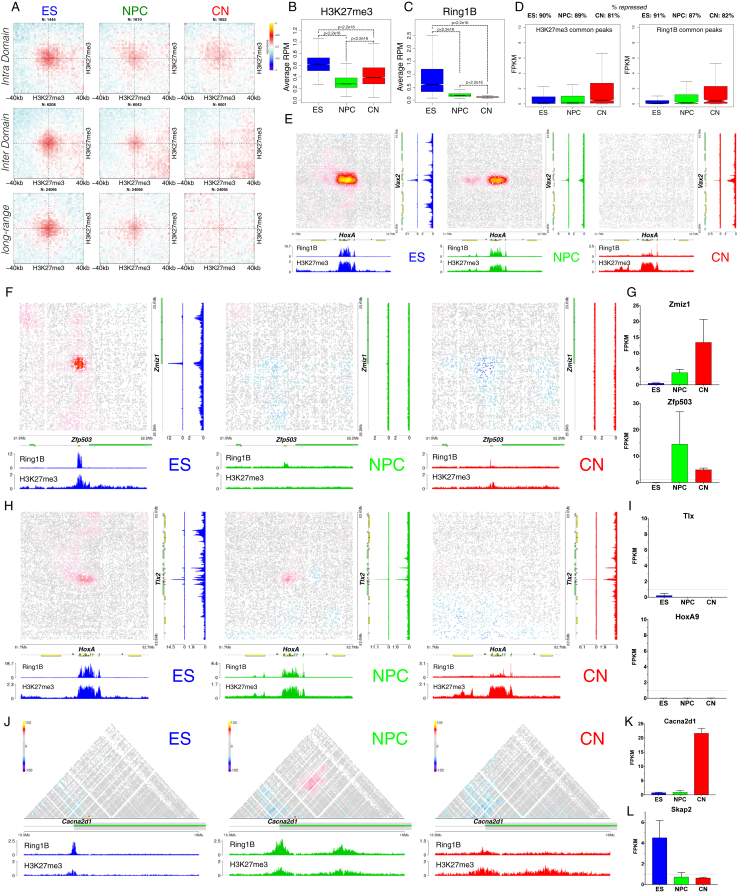
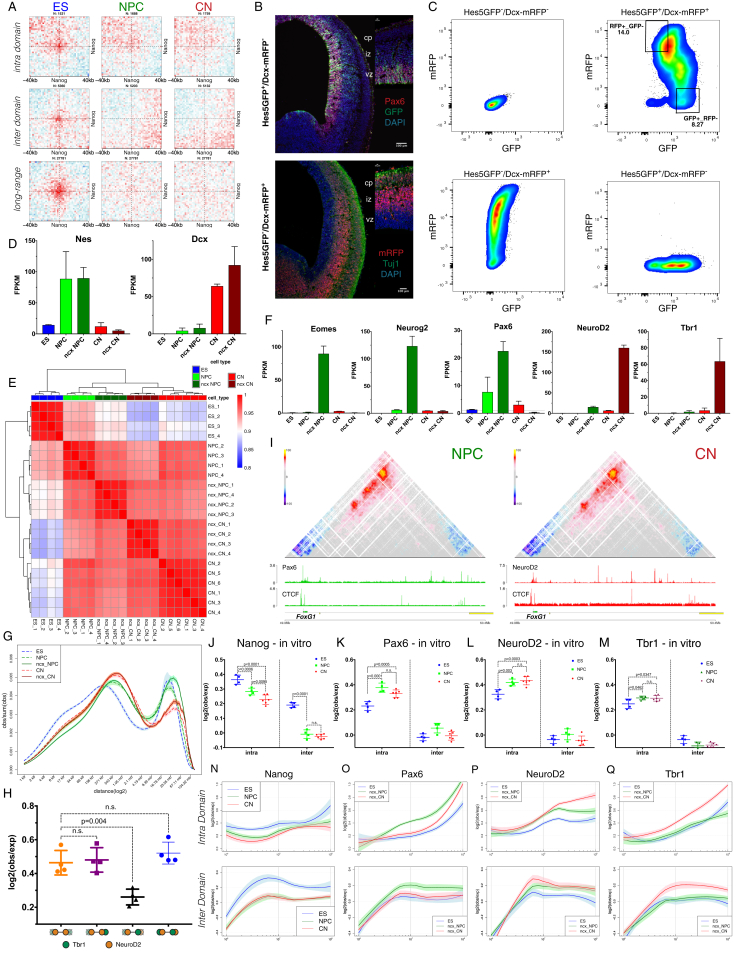
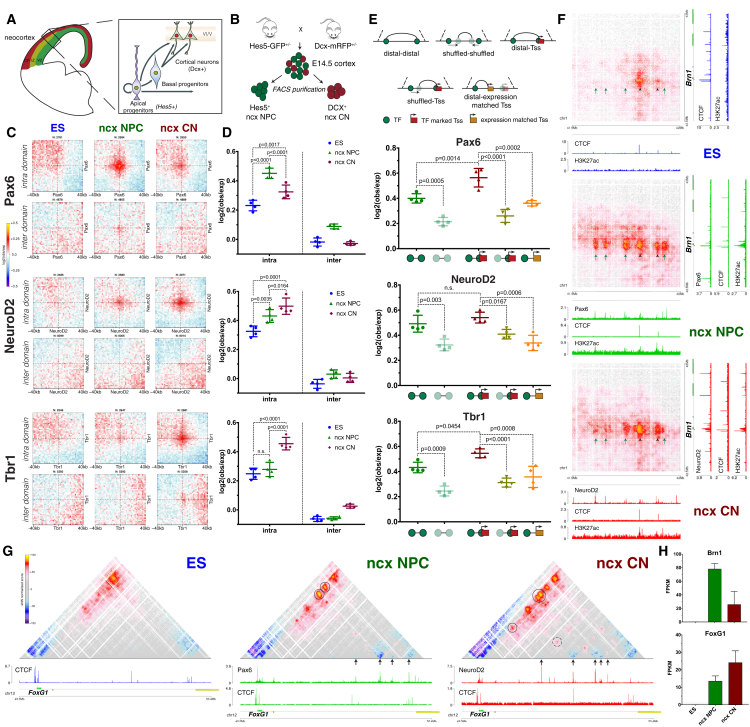
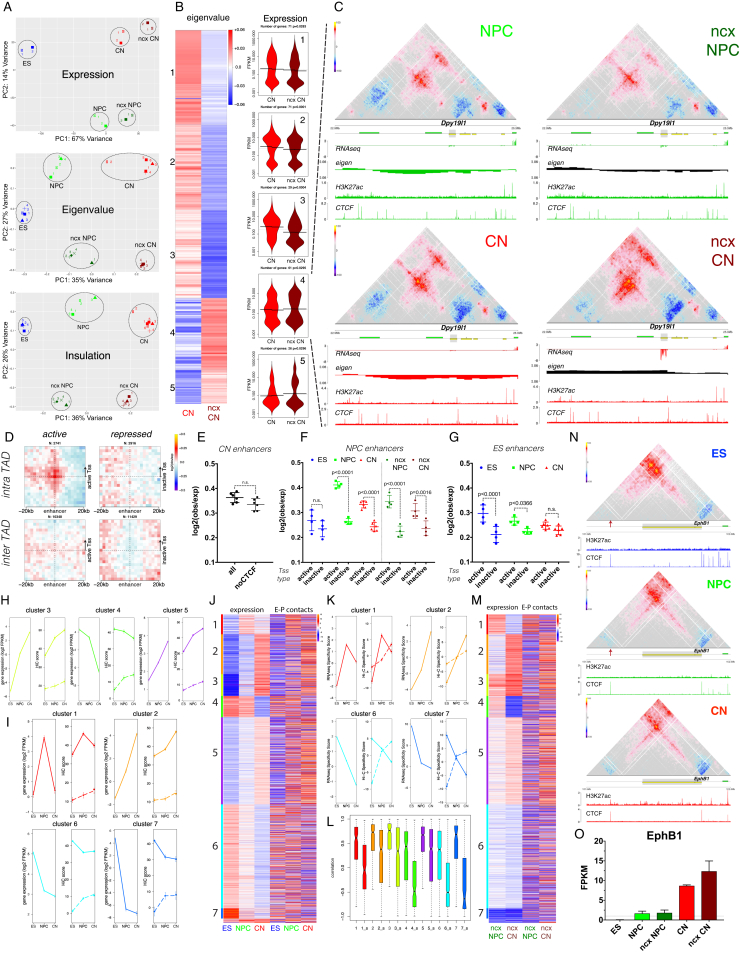
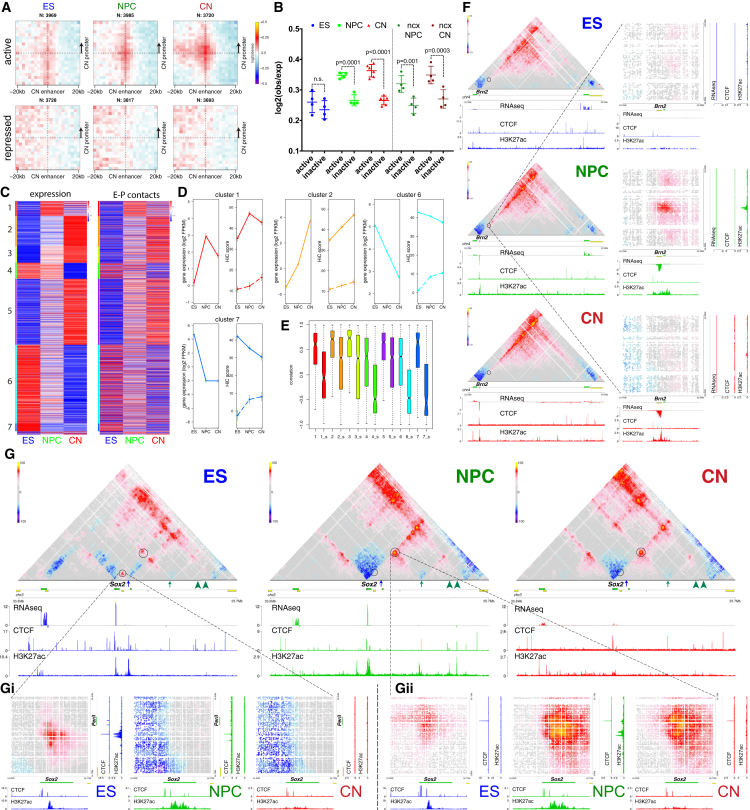
Chromosome conformation capture technologies have revealed important insights into genome folding. Yet, how spatial genome architecture is related to gene expression and cell fate remains unclear. We comprehensively mapped 3D chromatin organization during mouse neural differentiation in vitro and in vivo, generating the highest-resolution Hi-C maps available to date. We found that transcription is correlated with chromatin insulation and long-range interactions, but dCas9-mediated activation is insufficient for creating TAD boundaries de novo. Additionally, we discovered long-range contacts between gene bodies of exon-rich, active genes in all cell types. During neural differentiation, contacts between active TADs become less pronounced while inactive TADs interact more strongly. An extensive Polycomb network in stem cells is disrupted, while dynamic interactions between neural transcription factors appear in vivo. Finally, cell type-specific enhancer-promoter contacts are established concomitant to gene expression. This work shows that multiple factors influence the dynamics of chromatin interactions in development.
Keywords: 3D genome architecture; Hi-C; Polycomb; cortical development; enhancers; neural differentiation; transcription; transcription factors.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Bantignies F., Roure V., Comet I., Leblanc B., Schuettengruber B., Bonnet J., Tixier V., Mas A., Cavalli G. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 2011;144:214–226. - PubMed
-
- Basak O., Taylor V. Identification of self-replicating multipotent progenitors in the embryonic nervous system by high Notch activity and Hes5 expression. Eur. J. Neurosci. 2007;25:1006–1022. - PubMed
-
- Bibel M., Richter J., Schrenk K., Tucker K.L., Staiger V., Korte M., Goetz M., Barde Y.-A. Differentiation of mouse embryonic stem cells into a defined neuronal lineage. Nat. Neurosci. 2004;7:1003–1009. - PubMed
-
- Bonev B., Cavalli G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016;17:661–678. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
