

Acetaminophen Toxicity: Novel Insights Into Mechanisms and Future Perspectives
- PMID: 29054140
- PMCID: PMC5885144
- DOI: 10.3727/105221617X15084371374138
Acetaminophen Toxicity: Novel Insights Into Mechanisms and Future Perspectives
Abstract
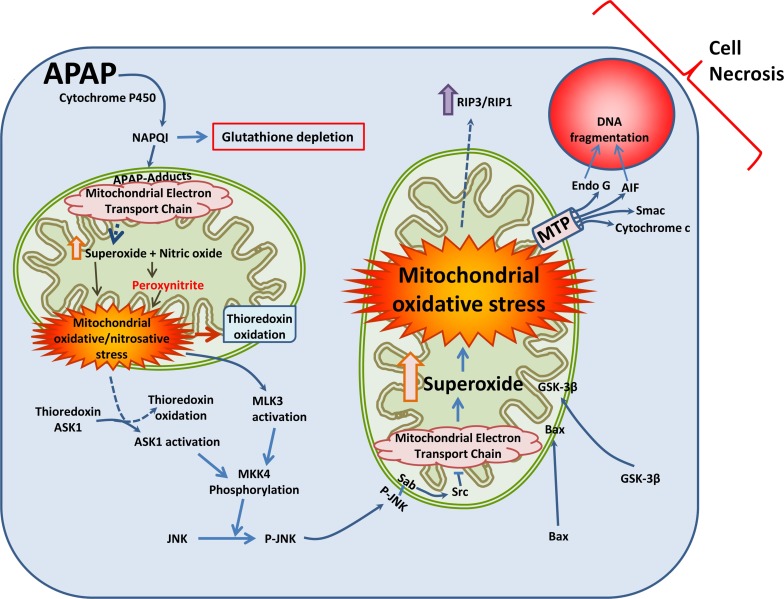
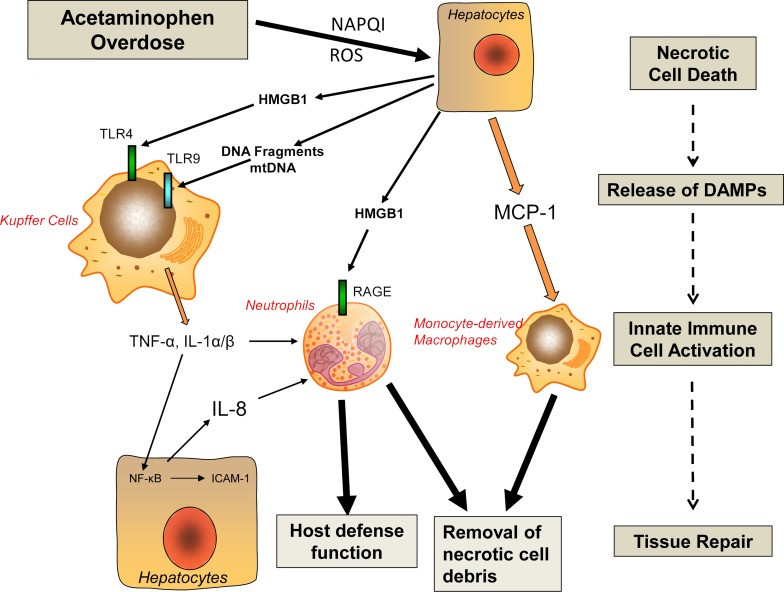
Acetaminophen (APAP) overdose is the most common cause of acute liver failure in the US, and decades of intense study of its pathogenesis resulted in the development of the antidote N-acetylcysteine, which facilitates scavenging of the reactive metabolite and is the only treatment in clinical use. However, the narrow therapeutic window of this intervention necessitates a better understanding of the intricacies of APAP-induced liver injury for the development of additional therapeutic approaches that can benefit late-presenting patients. More recent investigations into APAP hepatotoxicity have established the critical role of mitochondrial dysfunction in mediating liver injury as well as clarified mechanisms of APAP-induced hepatocyte cell death. Thus, it is now established that mitochondrial oxidative and nitrosative stress is a key mechanistic feature involved in downstream signaling after APAP overdose. The identification of specific mediators of necrotic cell death further establishes the regulated nature of APAP-induced hepatocyte cell death. In addition, the discovery of the role of mitochondrial dynamics and autophagy in APAP-induced liver injury provides additional insight into the elaborate cell signaling mechanisms involved in the pathogenesis of this important clinical problem. In spite of these new insights into the mechanisms of liver injury, significant controversy still exists on the role of innate immunity in APAP-induced hepatotoxicity.
Figures
References
-
- Lee WM. Acetaminophen-related acute liver failure in the United States. Hepatol Res. 2008;38(Suppl 1):S3–8. - PubMed
-
- Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiodt FV, Ostapowicz G, Shakil AO, Lee WM; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology 2005;42(6):1364–72. - PubMed
-
- Bernal W, Wendon J. Acute liver failure. N Engl J Med. 2013;369(26):2525–34. - PubMed
-
- Lancaster EM, Hiatt JR, Zarrinpar A. Acetaminophen hepatotoxicity: An updated review. Arch Toxicol. 2015;89(2):193–9. - PubMed
-
- Budnitz DS, Lovegrove MC, Crosby AE. Emergency department visits for overdoses of acetaminophen-containing products. Am J Prev Med. 2011;40(6):585–92. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical