

ATR/CHK1 inhibitors and cancer therapy
- PMID: 29054375
- PMCID: PMC5856582
- DOI: 10.1016/j.radonc.2017.09.043
ATR/CHK1 inhibitors and cancer therapy
Abstract
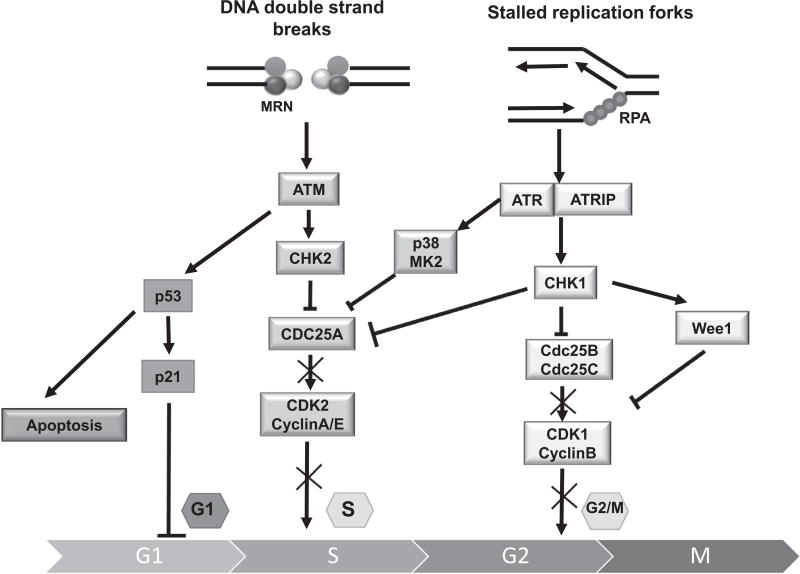
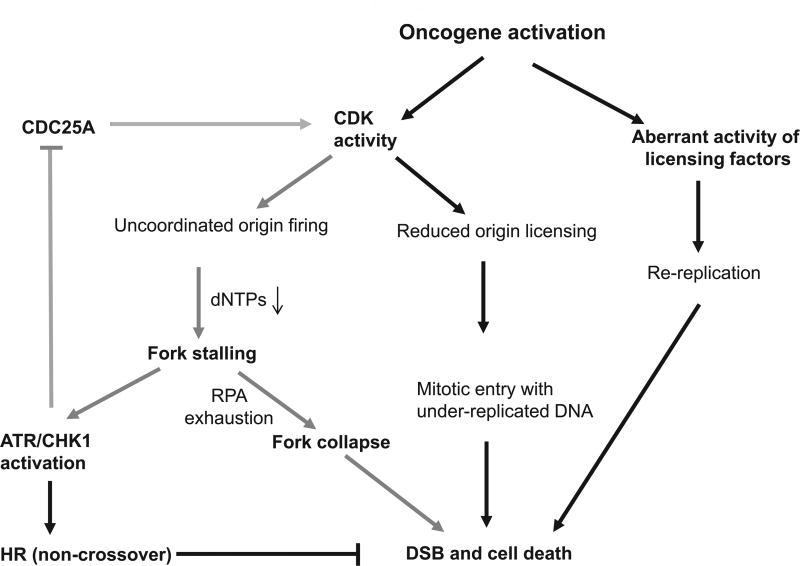
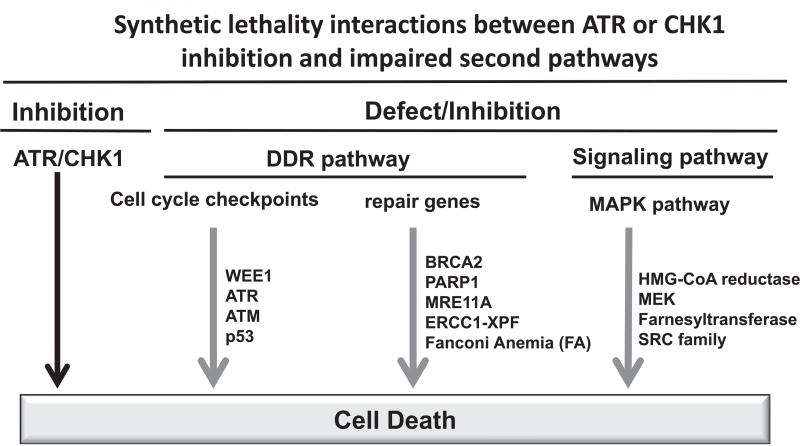
The cell cycle checkpoint proteins ataxia-telangiectasia-mutated-and-Rad3-related kinase (ATR) and its major downstream effector checkpoint kinase 1 (CHK1) prevent the entry of cells with damaged or incompletely replicated DNA into mitosis when the cells are challenged by DNA damaging agents, such as radiation therapy (RT) or chemotherapeutic drugs, that are the major modalities to treat cancer. This regulation is particularly evident in cells with a defective G1 checkpoint, a common feature of cancer cells, due to p53 mutations. In addition, ATR and/or CHK1 suppress replication stress (RS) by inhibiting excess origin firing, particularly in cells with activated oncogenes. Those functions of ATR/CHK1 make them ideal therapeutic targets. ATR/CHK1 inhibitors have been developed and are currently used either as single agents or paired with radiotherapy or a variety of genotoxic chemotherapies in preclinical and clinical studies. Here, we review the status of the development of ATR and CHK1 inhibitors. We also discuss the potential mechanisms by which ATR and CHK1 inhibition induces cell killing in the presence or absence of exogenous DNA damaging agents, such as RT and chemotherapeutic agents. Lastly, we discuss synthetic lethality interactions between the inhibition of ATR/CHK1 and defects in other DNA damage response (DDR) pathways/genes.
Keywords: ATR; CHK1; Cell cycle checkpoints; DNA damage response; DNA replication stress; Synthetic lethality.
Copyright © 2017 Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–17. - PubMed
-
- De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169–85. - PubMed
-
- Lavin MF. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. 2007;26:7749–58. - PubMed
-
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
