

Comprehensive Analysis of Hypermutation in Human Cancer
- PMID: 29056344
- PMCID: PMC5849393
- DOI: 10.1016/j.cell.2017.09.048
Comprehensive Analysis of Hypermutation in Human Cancer
Abstract
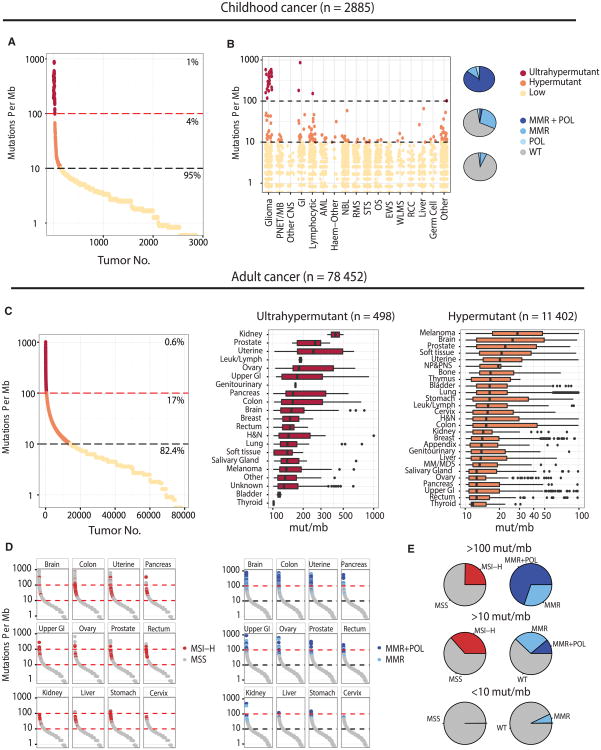
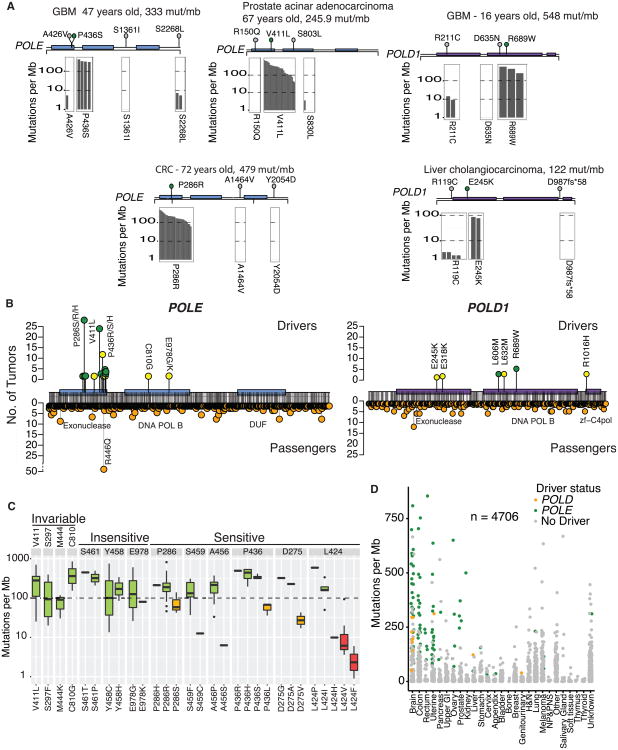
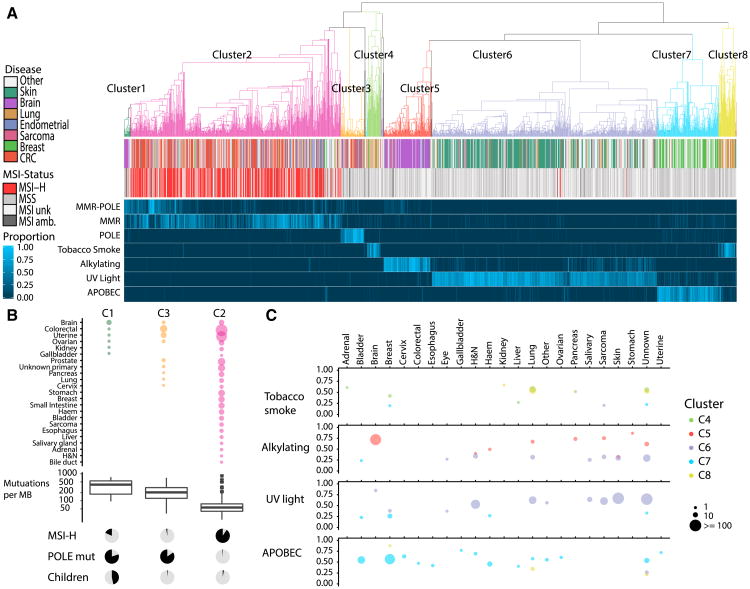
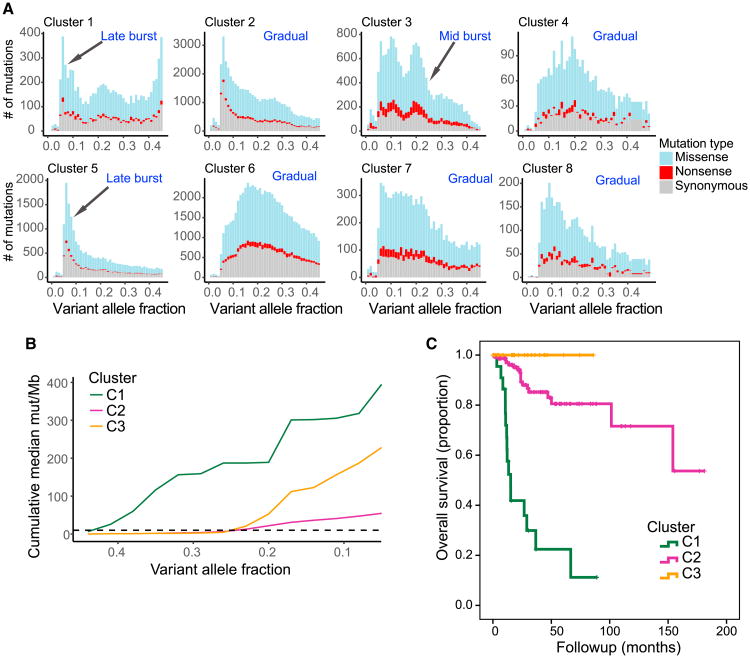
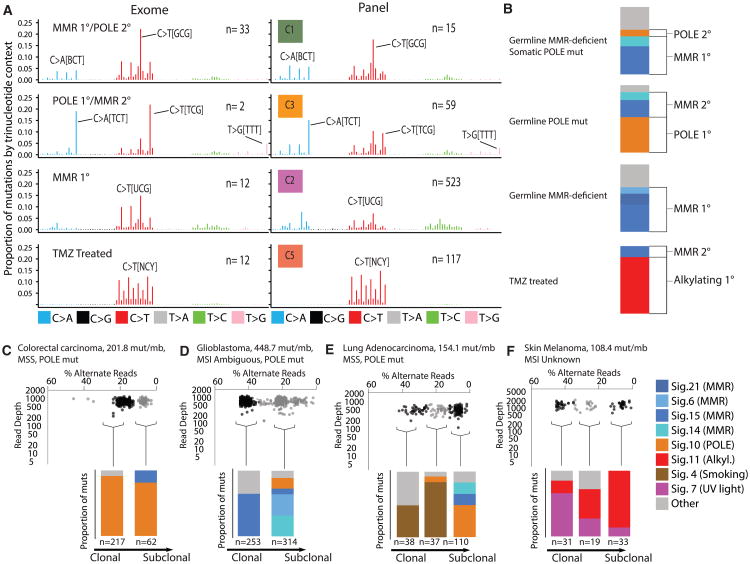
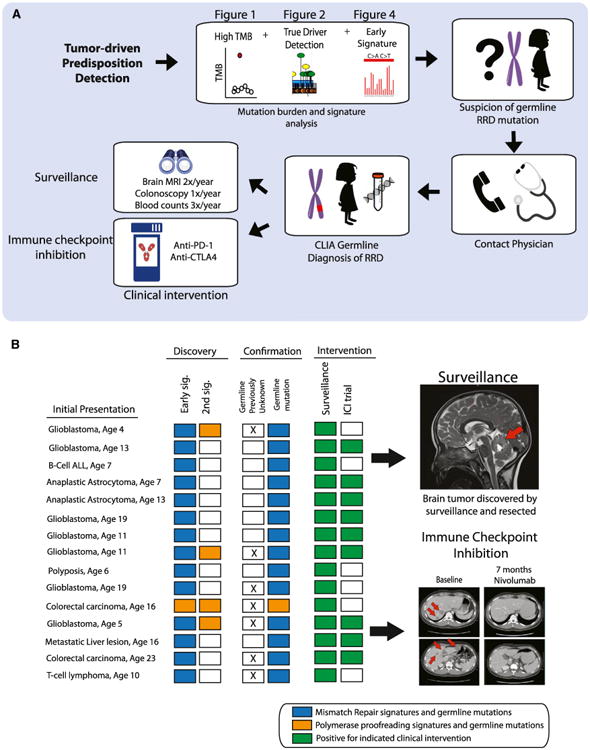
We present an extensive assessment of mutation burden through sequencing analysis of >81,000 tumors from pediatric and adult patients, including tumors with hypermutation caused by chemotherapy, carcinogens, or germline alterations. Hypermutation was detected in tumor types not previously associated with high mutation burden. Replication repair deficiency was a major contributing factor. We uncovered new driver mutations in the replication-repair-associated DNA polymerases and a distinct impact of microsatellite instability and replication repair deficiency on the scale of mutation load. Unbiased clustering, based on mutational context, revealed clinically relevant subgroups regardless of the tumors' tissue of origin, highlighting similarities in evolutionary dynamics leading to hypermutation. Mutagens, such as UV light, were implicated in unexpected cancers, including sarcomas and lung tumors. The order of mutational signatures identified previous treatment and germline replication repair deficiency, which improved management of patients and families. These data will inform tumor classification, genetic testing, and clinical trial design.
Keywords: DNA repair; DNA replication; cancer genomics; cancer predisposition; hypermutation; immune checkpoint inhibitors; mismatch repair; mutator.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Cancer genomics: The driving force of cancer evolution.Nat Rev Genet. 2017 Dec;18(12):703. doi: 10.1038/nrg.2017.95. Epub 2017 Nov 7. Nat Rev Genet. 2017. PMID: 29109522 No abstract available.
-
Everybody In! No Bouncers at Tumor Gates.Trends Genet. 2018 Feb;34(2):85-87. doi: 10.1016/j.tig.2017.12.006. Epub 2017 Dec 23. Trends Genet. 2018. PMID: 29277455
References
-
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. - PMC - PubMed
-
- Amayiri N, Tabori U, Campbell B, Bakry D, Aronson M, Durno C, Rakopoulos P, Malkin D, Qaddoumi I, Musharbash A, et al. BMMRD Consortium. High frequency of mismatch repair deficiency among pediatric high grade gliomas in Jordan. Int J Cancer. 2016;138:380–385. - PubMed
-
- Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34:2206–2211. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
