

Universal Patterns of Selection in Cancer and Somatic Tissues
- PMID: 29056346
- PMCID: PMC5720395
- DOI: 10.1016/j.cell.2017.09.042
Universal Patterns of Selection in Cancer and Somatic Tissues
Erratum in
-
Universal Patterns of Selection in Cancer and Somatic Tissues.Cell. 2018 Jun 14;173(7):1823. doi: 10.1016/j.cell.2018.06.001. Cell. 2018. PMID: 29906452 Free PMC article. No abstract available.
Abstract
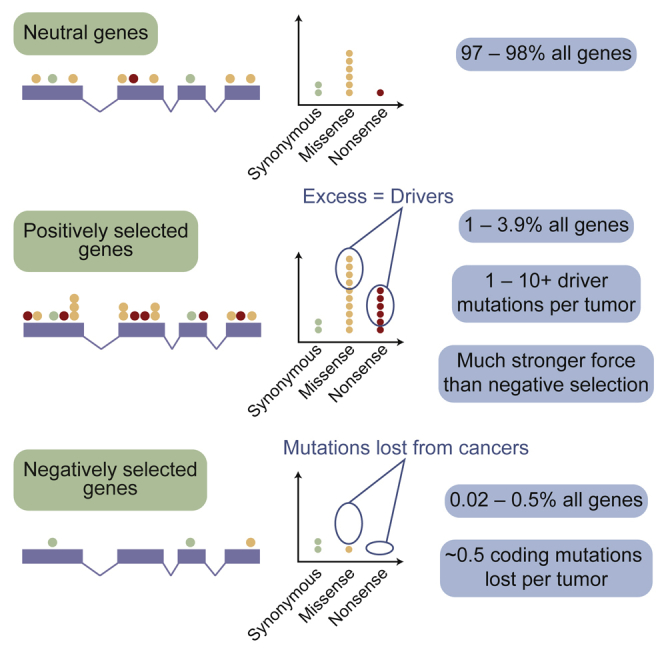
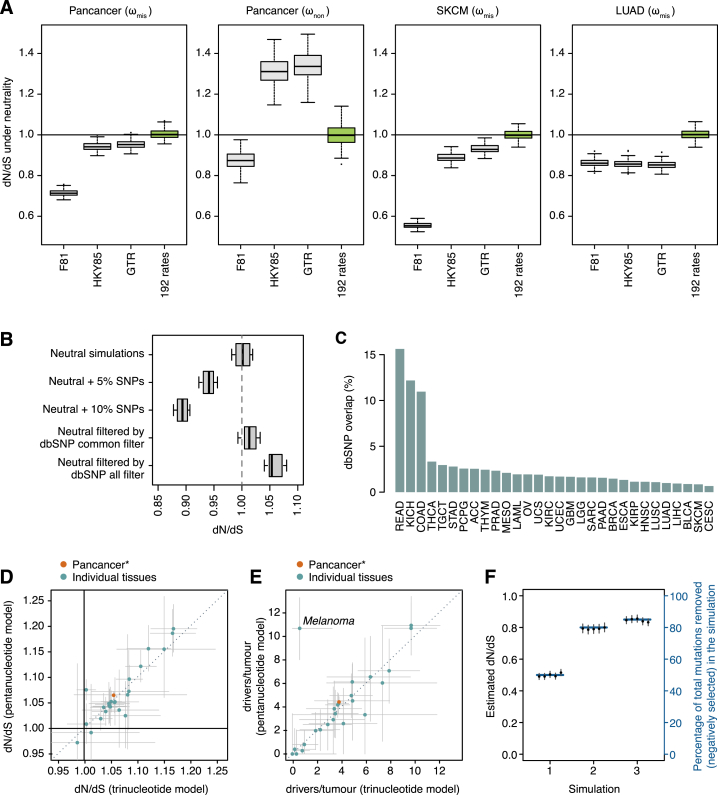
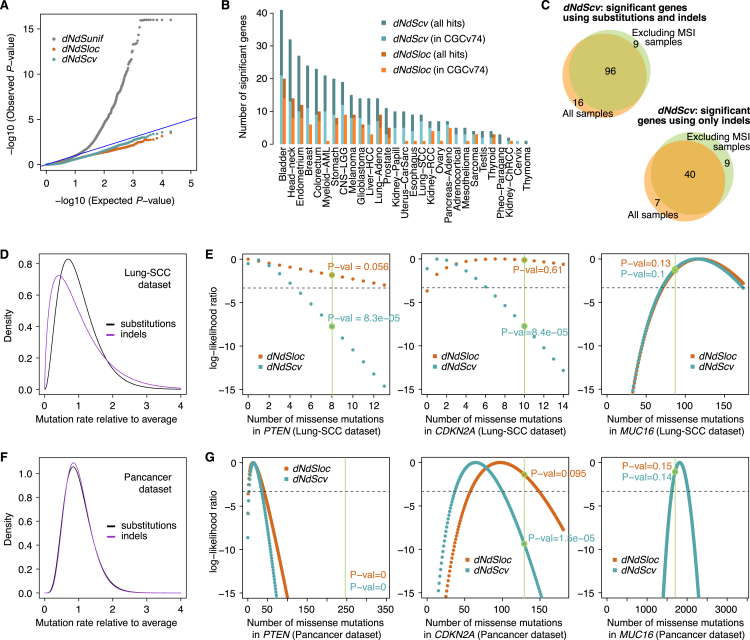
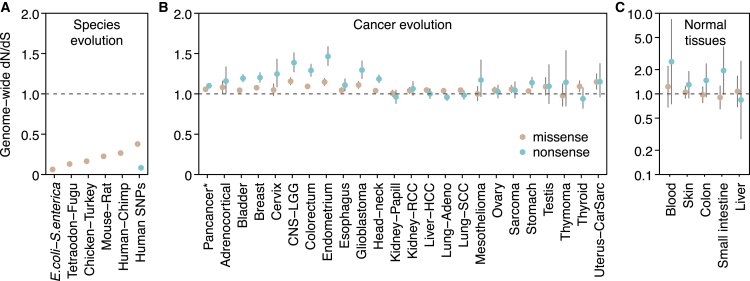
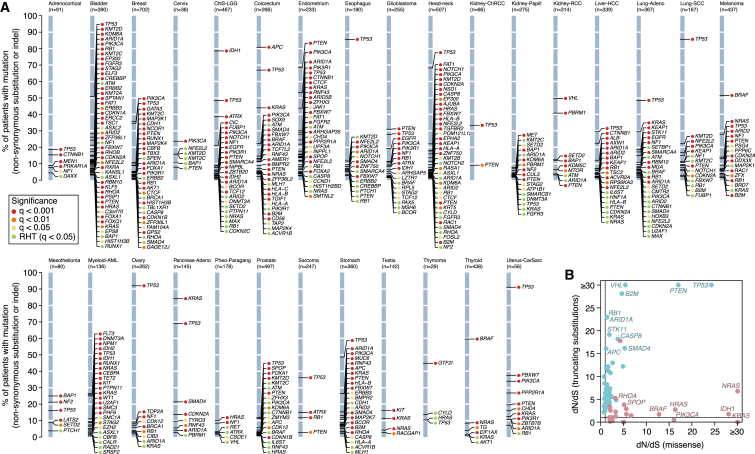
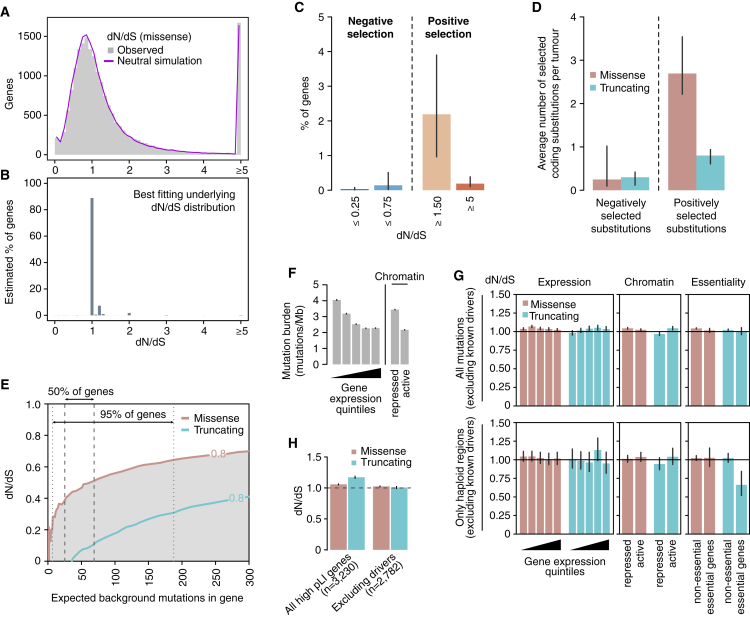
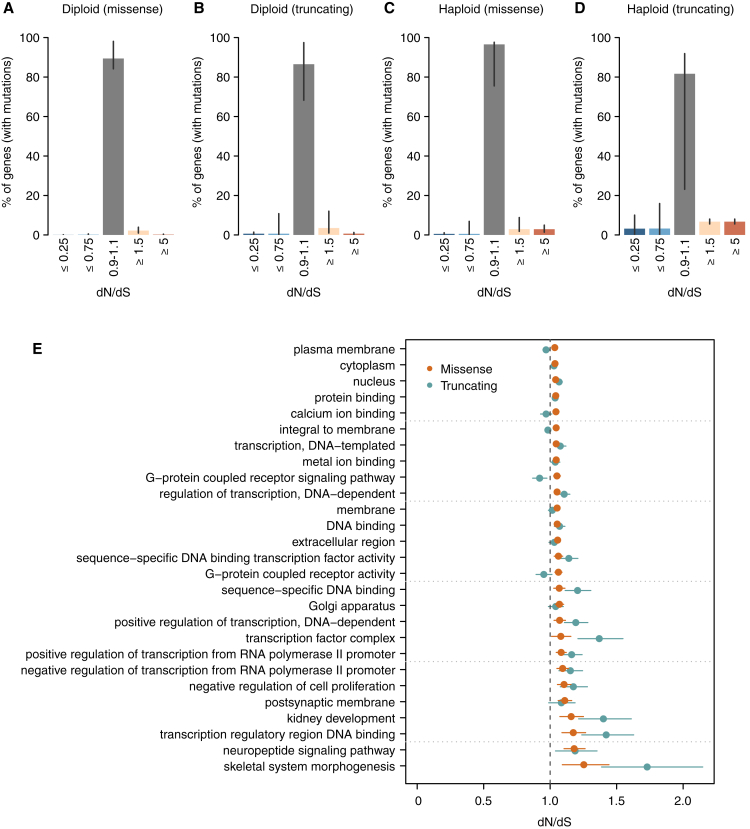
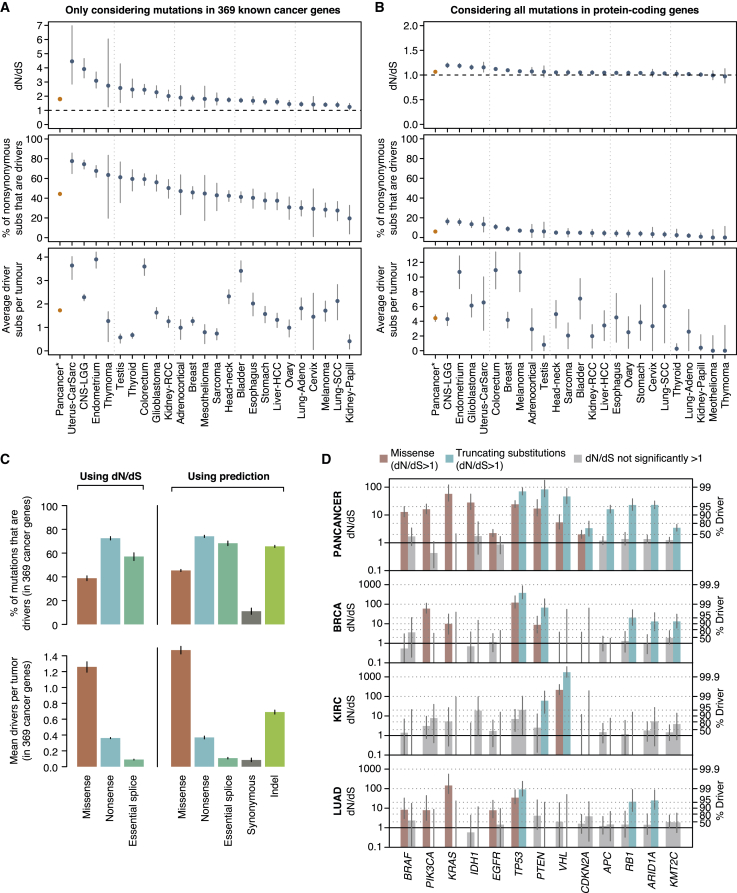
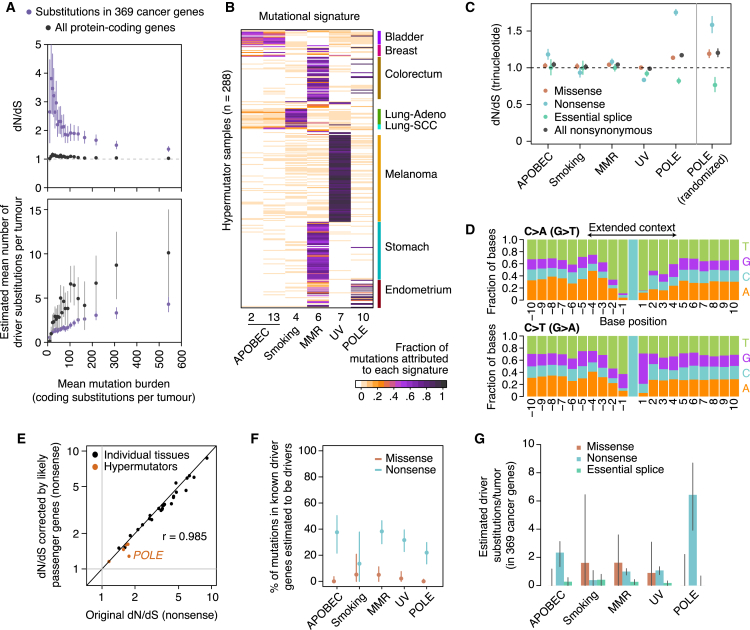
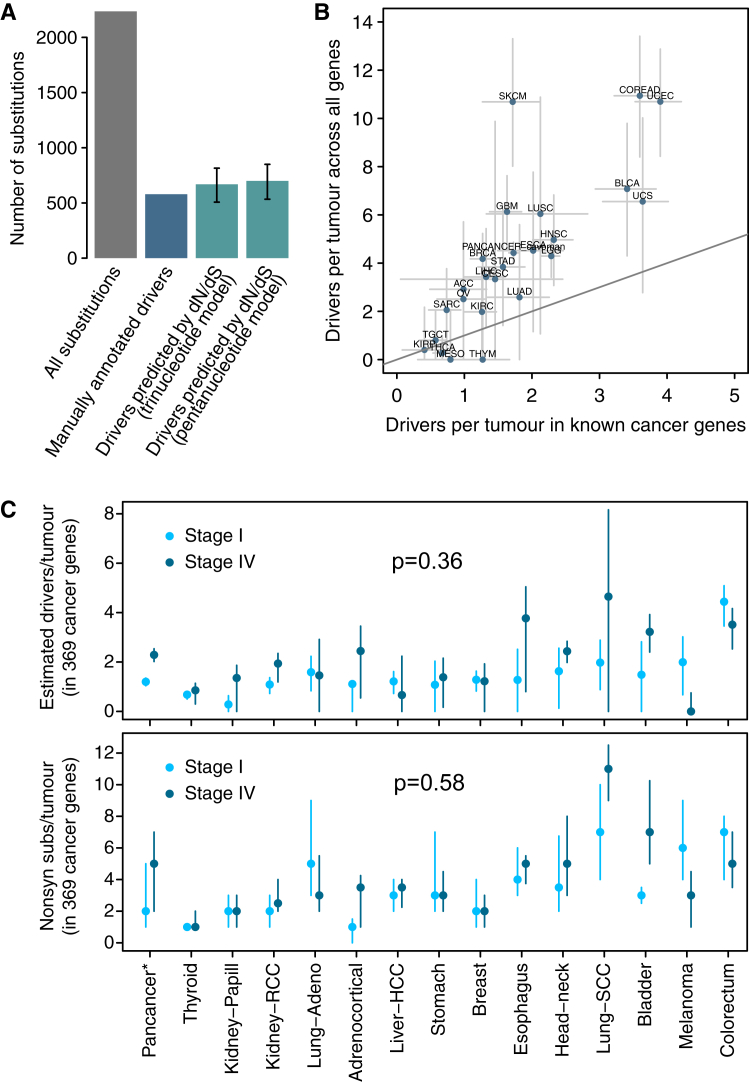
Cancer develops as a result of somatic mutation and clonal selection, but quantitative measures of selection in cancer evolution are lacking. We adapted methods from molecular evolution and applied them to 7,664 tumors across 29 cancer types. Unlike species evolution, positive selection outweighs negative selection during cancer development. On average, <1 coding base substitution/tumor is lost through negative selection, with purifying selection almost absent outside homozygous loss of essential genes. This allows exome-wide enumeration of all driver coding mutations, including outside known cancer genes. On average, tumors carry ∼4 coding substitutions under positive selection, ranging from <1/tumor in thyroid and testicular cancers to >10/tumor in endometrial and colorectal cancers. Half of driver substitutions occur in yet-to-be-discovered cancer genes. With increasing mutation burden, numbers of driver mutations increase, but not linearly. We systematically catalog cancer genes and show that genes vary extensively in what proportion of mutations are drivers versus passengers.
Keywords: cancer; evolution; genomics; mutations; selection.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
Cancer genomics: The driving force of cancer evolution.Nat Rev Genet. 2017 Dec;18(12):703. doi: 10.1038/nrg.2017.95. Epub 2017 Nov 7. Nat Rev Genet. 2017. PMID: 29109522 No abstract available.
-
Cancer Evolution: No Room for Negative Selection.Cell. 2017 Nov 16;171(5):987-989. doi: 10.1016/j.cell.2017.10.039. Cell. 2017. PMID: 29149612
-
Everybody In! No Bouncers at Tumor Gates.Trends Genet. 2018 Feb;34(2):85-87. doi: 10.1016/j.tig.2017.12.006. Epub 2017 Dec 23. Trends Genet. 2018. PMID: 29277455
References
-
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
