

PRRT2 deficiency induces paroxysmal kinesigenic dyskinesia by regulating synaptic transmission in cerebellum
- PMID: 29056747
- PMCID: PMC5752836
- DOI: 10.1038/cr.2017.128
PRRT2 deficiency induces paroxysmal kinesigenic dyskinesia by regulating synaptic transmission in cerebellum
Abstract
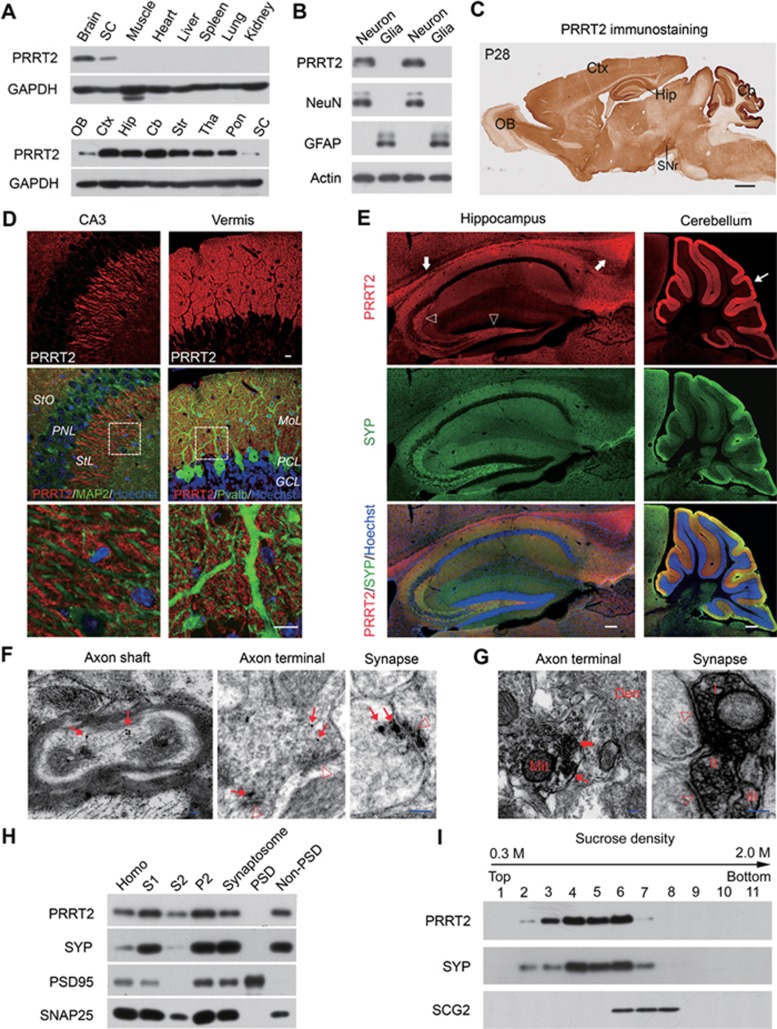
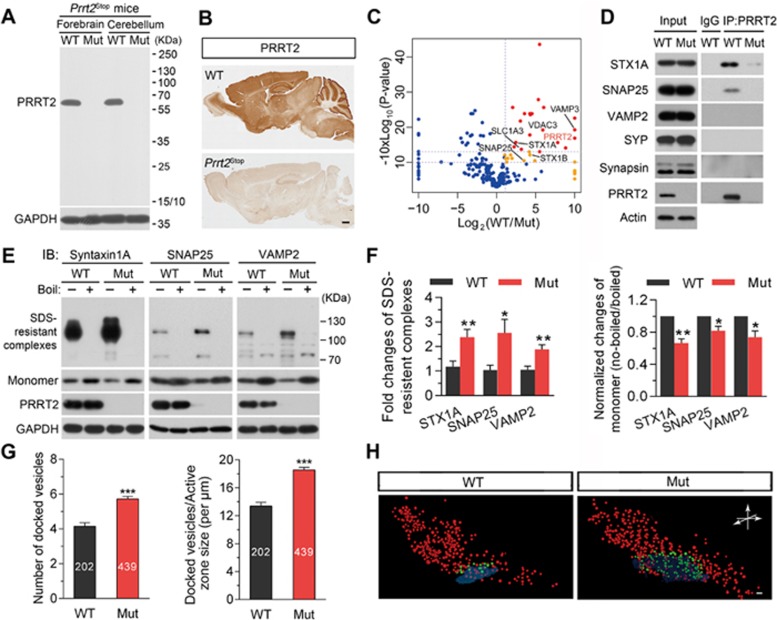
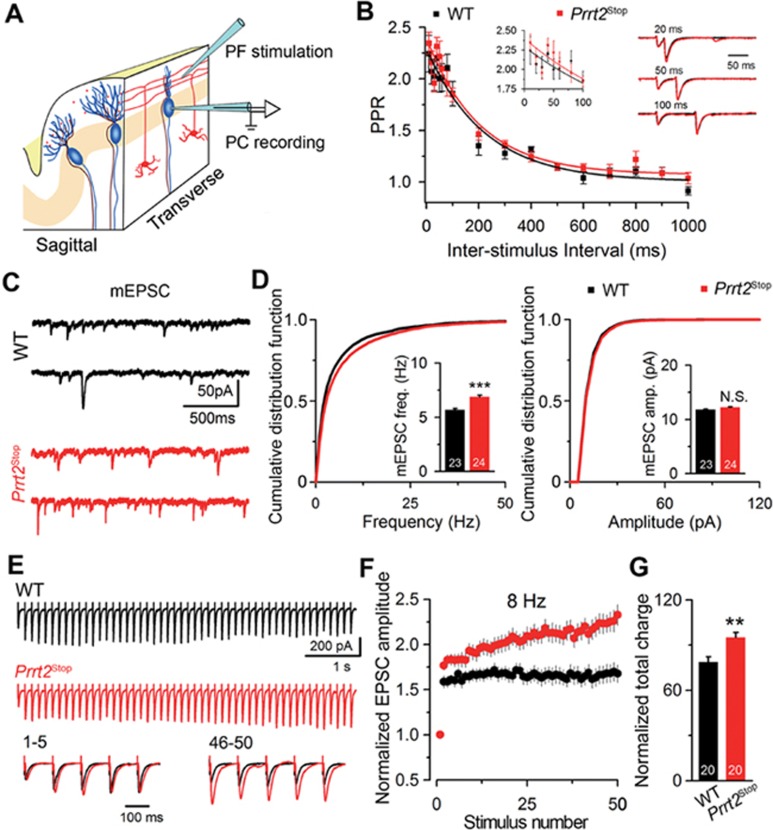
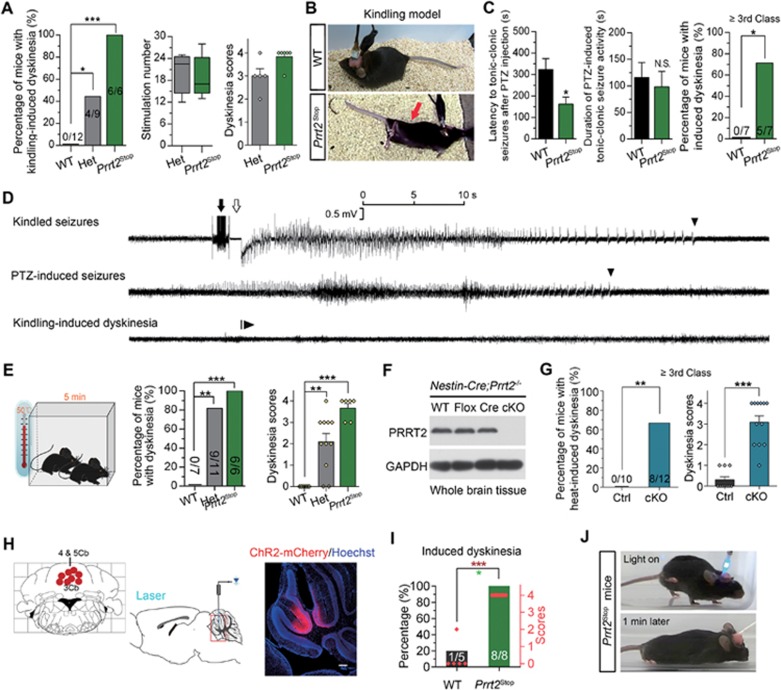
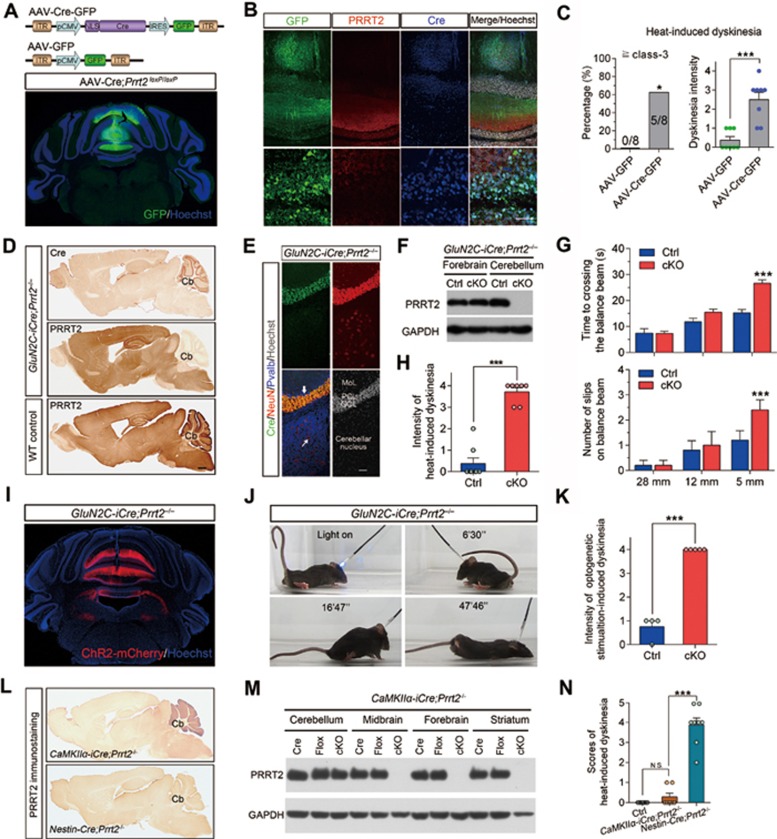
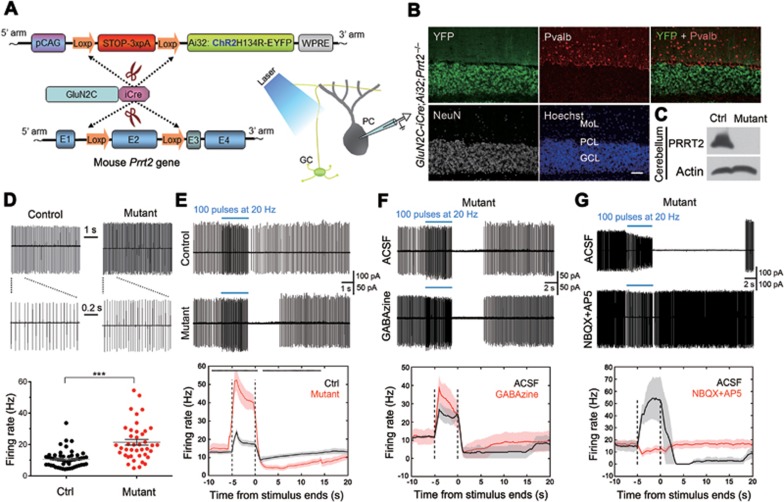
Mutations in the proline-rich transmembrane protein 2 (PRRT2) are associated with paroxysmal kinesigenic dyskinesia (PKD) and several other paroxysmal neurological diseases, but the PRRT2 function and pathogenic mechanisms remain largely obscure. Here we show that PRRT2 is a presynaptic protein that interacts with components of the SNARE complex and downregulates its formation. Loss-of-function mutant mice showed PKD-like phenotypes triggered by generalized seizures, hyperthermia, or optogenetic stimulation of the cerebellum. Mutant mice with specific PRRT2 deletion in cerebellar granule cells (GCs) recapitulate the behavioral phenotypes seen in Prrt2-null mice. Furthermore, recording made in cerebellar slices showed that optogenetic stimulation of GCs results in transient elevation followed by suppression of Purkinje cell firing. The anticonvulsant drug carbamazepine used in PKD treatment also relieved PKD-like behaviors in mutant mice. Together, our findings identify PRRT2 as a novel regulator of the SNARE complex and provide a circuit mechanism underlying the PRRT2-related behaviors.
Figures
Comment in
-
PRRT2-dependent dyskinesia: cerebellar, paroxysmal and persistent.Cell Res. 2018 Jan;28(1):3-4. doi: 10.1038/cr.2017.142. Epub 2017 Nov 17. Cell Res. 2018. PMID: 29148542 Free PMC article.
References
-
- Bhatia KP. Paroxysmal dyskinesias. Mov Disord 2011; 26:1157–1165. - PubMed
-
- Ebrahimi-Fakhari D, Saffari A, Westenberger A, Klein C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain 2015; 138:3476–3495 - PubMed
-
- Chen WJ, Lin Y, Xiong ZQ, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet 2011; 43:1252–1255. - PubMed
-
- Meneret A, Gaudebout C, Riant F, et al. PRRT2 mutations and paroxysmal disorders. Eur J Neurol 2013; 20:872–878. - PubMed
-
- Schubert J, Paravidino R, Becker F, et al. PRRT2 mutations are the major cause of benign familial infantile seizures. Hum Mutat 2012; 33:1439–1443. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
