

De novo chromosome 7q36.1q36.2 triplication in a child with developmental delay, growth failure, distinctive facial features, and multiple congenital anomalies: a case report
- PMID: 29061174
- PMCID: PMC5654040
- DOI: 10.1186/s12881-017-0482-8
De novo chromosome 7q36.1q36.2 triplication in a child with developmental delay, growth failure, distinctive facial features, and multiple congenital anomalies: a case report
Abstract
Background: Studying human genome using chromosomal microarrays has significantly improved the accuracy and yield of diagnosing genomic disorders. Chromosome 7q36 deletions and duplications are rare genomic disorders that have been reported in a limited number of children with developmental delay, growth retardation, and congenital malformation. Altered dosage of SHH and HLXB9, both located in 7q36.3, is believed to play roles in the phenotypes associated with these rearrangements. In this report we describe a child with 7q36.1q36.2 triplication that is proximal to the 7q36.3 region. In addition to the clinical description, we discuss the genes located in the triplicated region.
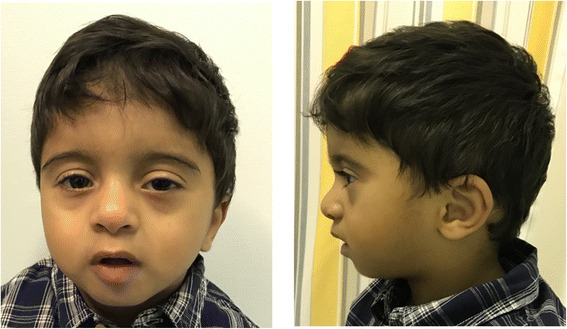
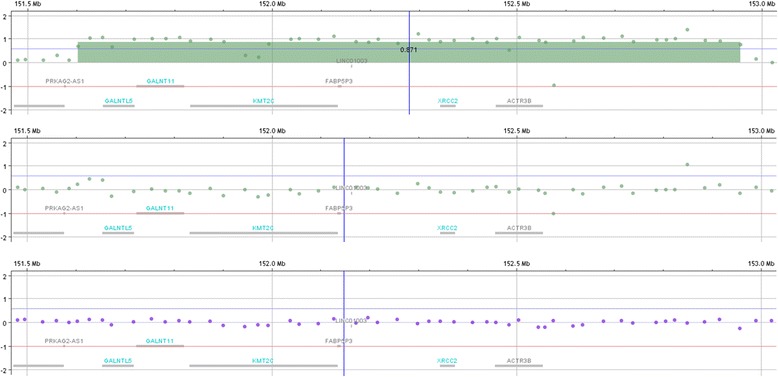
Case presentation: We report a 22 month old male child with a de novo 1.35 Mb triplication at 7q36.1q36.2. His prenatal course was complicated by oligohydramnios, intrauterine growth restriction, and decreased fetal movement. Hypotonia, respiratory distress, and feeding difficulty were observed in the neonatal period. He also had developmental delay, cardiovascular malformation, growth failure with microcephaly, short stature, and underweight, sensorineural hearing loss, myopia, astigmatism, cryptorchidism, hypospadias, microphallus, lower extremity length discrepancy, bifid uvula, single palmer creases, and distinctive facial features including straight eyebrows, ptosis, up-slanted palpebral fissures, broad nasal bridge, low-set and posteriorly rotated ears, small mouth with thick lower lip, microretrognathia, and high-arched palate.
Conclusions: The child presented here had developmental delay, distinctive facial features, multiple congenital anomalies, and 7q36.1q36.2 triplication. This triplication, which was found to be de novo, has not been previously described and is believed to result in the observed phenotype. The triplicated region harbors the GALNTL5, GALNT11, KMT2C, XRCC2, and ACTR3B genes. GALNT11 encodes a membrane-bound polypeptide N-acetylgalactosaminyltransferase that can O-glycosylate NOTCH1 leading to the activation of the Notch signaling pathway. Therefore, increased GALNT11 dosage can potentially alter the Notch signaling pathway explaining the pathogenicity of 7q36 triplication. Studying further cases with similar genomic rearrangements is needed to make final conclusions about the pathogenicity of this triplication.
Keywords: Chromosomal disorders; Chromosomal microarray; Chromosome 7q36; Genomic rearrangements.
Conflict of interest statement
Ethics approval and consent to participate
The study was approved by Al-Ain Medical District Human Research Ethics Committee (15/89-CRD 416/15). Informed consent was obtained from parents.
Consent for publication
Informed consent was obtained from parents who consented to publish the images as well as the medical information supplied in this case report.
Competing interests
DA-H is an employee of PreventionGenetics, LLC.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006. - DOI - PMC - PubMed
-
- Alabdullatif MA, Al Dhaibani MA, Khassawneh MY, El-Hattab AW. Chromosomal microarray in a highly consanguineous population: diagnostic yield, utility of regions of homozygosity, and novel mutations. Clin Genet. 2017;91(4):616–22. - PubMed
-
- Xiang B, Zhu H, Shen Y, Miller DT, Lu K, Hu X, et al. Genome-wide oligonucleotide array comparative genomic hybridization for etiological diagnosis of mental retardation: a multicenter experience of 1499 clinical cases. J Mol Diagn. 2010;12(2):204–212. doi: 10.2353/jmoldx.2010.090115. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
