

Structure of human lysosomal acid α-glucosidase-a guide for the treatment of Pompe disease
- PMID: 29061980
- PMCID: PMC5653652
- DOI: 10.1038/s41467-017-01263-3
Structure of human lysosomal acid α-glucosidase-a guide for the treatment of Pompe disease
Abstract
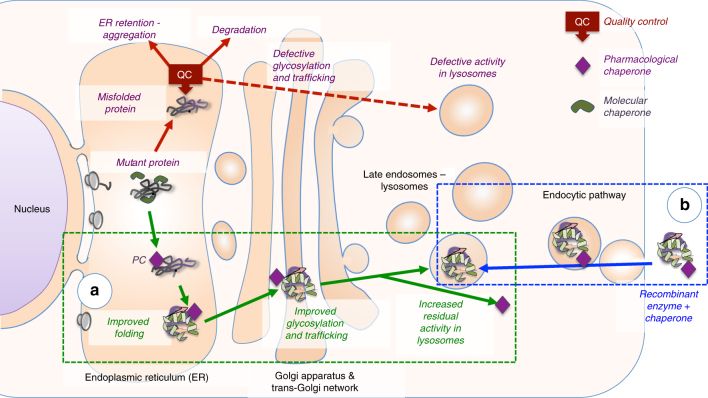
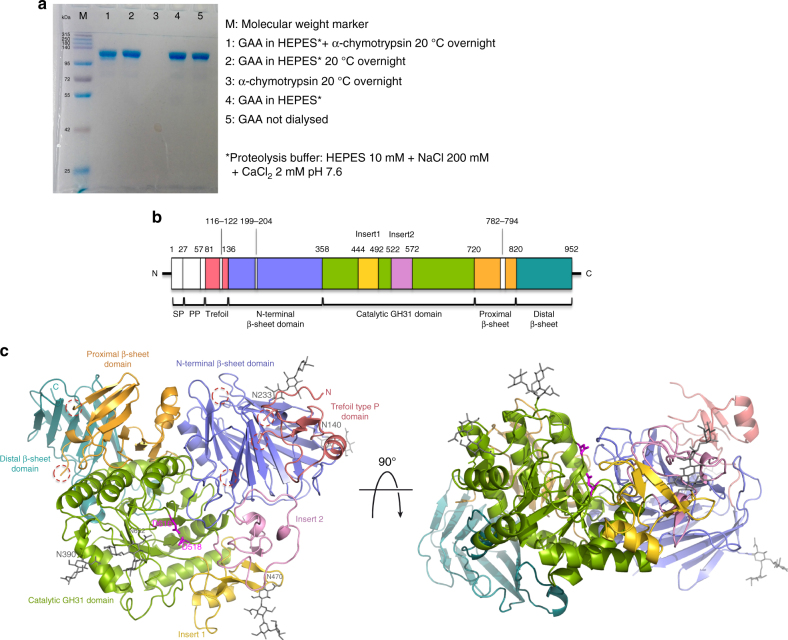
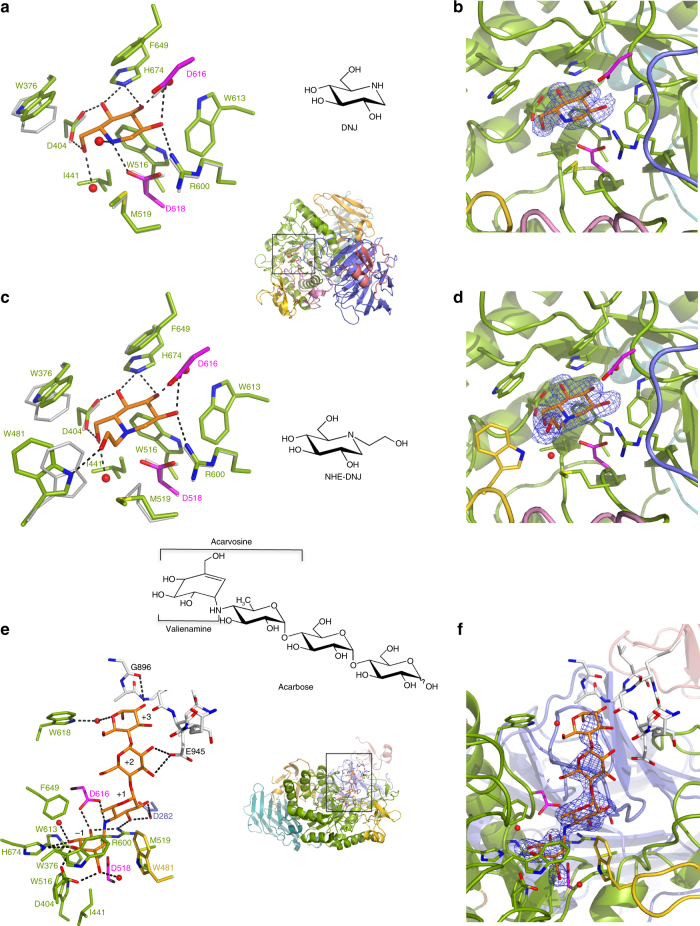
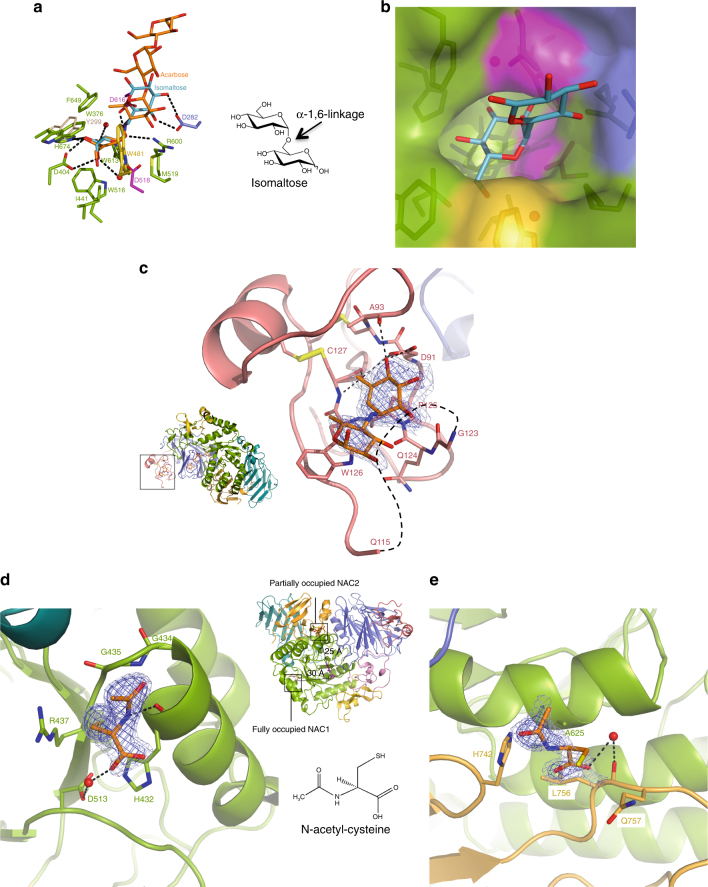
Pompe disease, a rare lysosomal storage disease caused by deficiency of the lysosomal acid α-glucosidase (GAA), is characterized by glycogen accumulation, triggering severe secondary cellular damage and resulting in progressive motor handicap and premature death. Numerous disease-causing mutations in the gaa gene have been reported, but the structural effects of the pathological variants were unknown. Here we present the high-resolution crystal structures of recombinant human GAA (rhGAA), the standard care of Pompe disease. These structures portray the unbound form of rhGAA and complexes thereof with active site-directed inhibitors, providing insight into substrate recognition and the molecular framework for the rationalization of the deleterious effects of disease-causing mutations. Furthermore, we report the structure of rhGAA in complex with the allosteric pharmacological chaperone N-acetylcysteine, which reveals the stabilizing function of this chaperone at the structural level.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
