

A Brain Penetrant Mutant IDH1 Inhibitor Provides In Vivo Survival Benefit
- PMID: 29062039
- PMCID: PMC5653818
- DOI: 10.1038/s41598-017-14065-w
A Brain Penetrant Mutant IDH1 Inhibitor Provides In Vivo Survival Benefit
Abstract
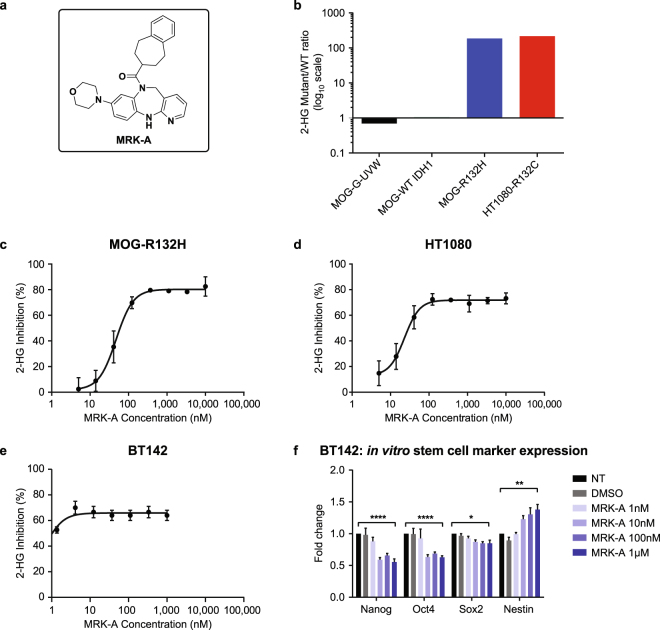
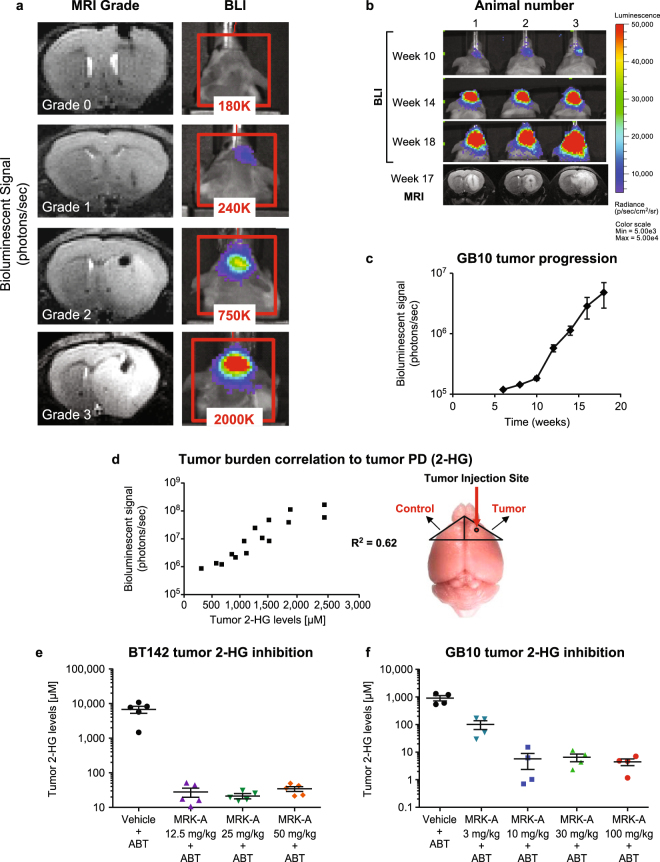
Mutations in IDH1 are highly prevalent in human glioma. First line treatment is radiotherapy, which many patients often forego to avoid treatment-associated morbidities. The high prevalence of IDH1 mutations in glioma highlights the need for brain-penetrant IDH1 mutant-selective inhibitors as an alternative therapeutic option. Here, we have explored the utility of such an inhibitor in IDH1 mutant patient-derived models to assess the potential therapeutic benefits associated with intracranial 2-HG inhibition. Treatment of mutant IDH1 cell line models led to a decrease in intracellular 2-HG levels both in vitro and in vivo. Interestingly, inhibition of 2-HG production had no effect on in vitro IDH1 mutant glioma cell proliferation. In contrast, IDH1 mutant-selective inhibitors provided considerable survival benefit in vivo. However, even with near complete inhibition of intratumoral 2-HG production, not all mutant glioma models responded to treatment. The results suggest that disruption of 2-HG production with brain-penetrant inhibitors in IDH1 mutant gliomas may have substantial patient benefit.
Conflict of interest statement
The study was sponsored by Merck & Co., Inc., Kenilworth, New Jersey, USA. J. Kopinja, R.S. Sevilla, D. Levitan, D. Dai, A. Vanko, E. Spooner, C. Ware, R. Forget, K. Hu, A. Kral, P. Spacciapoli, R. Kennan, L. Jayaraman, V. Pucci, S. Perera, W. Zhang, C. Fischer, and M.H. Lam are current or former employees of Merck Sharp & Dohme, Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, USA and may own stock or stock options in the company.
Figures



References
-
- Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013;3:730–741. doi: 10.1158/2159-8290.CD-13-0083. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
