

Genetic Variants Associated with Episodic Ataxia in Korea
- PMID: 29062094
- PMCID: PMC5653837
- DOI: 10.1038/s41598-017-14254-7
Genetic Variants Associated with Episodic Ataxia in Korea
Abstract
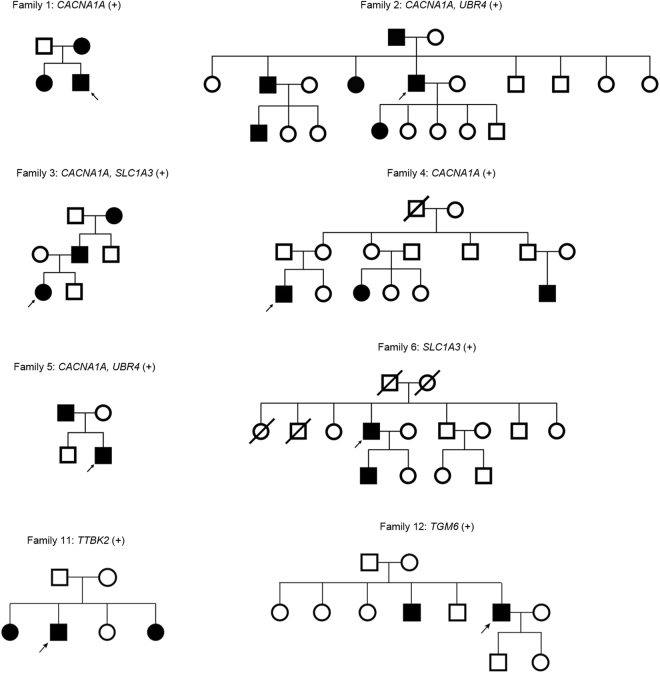
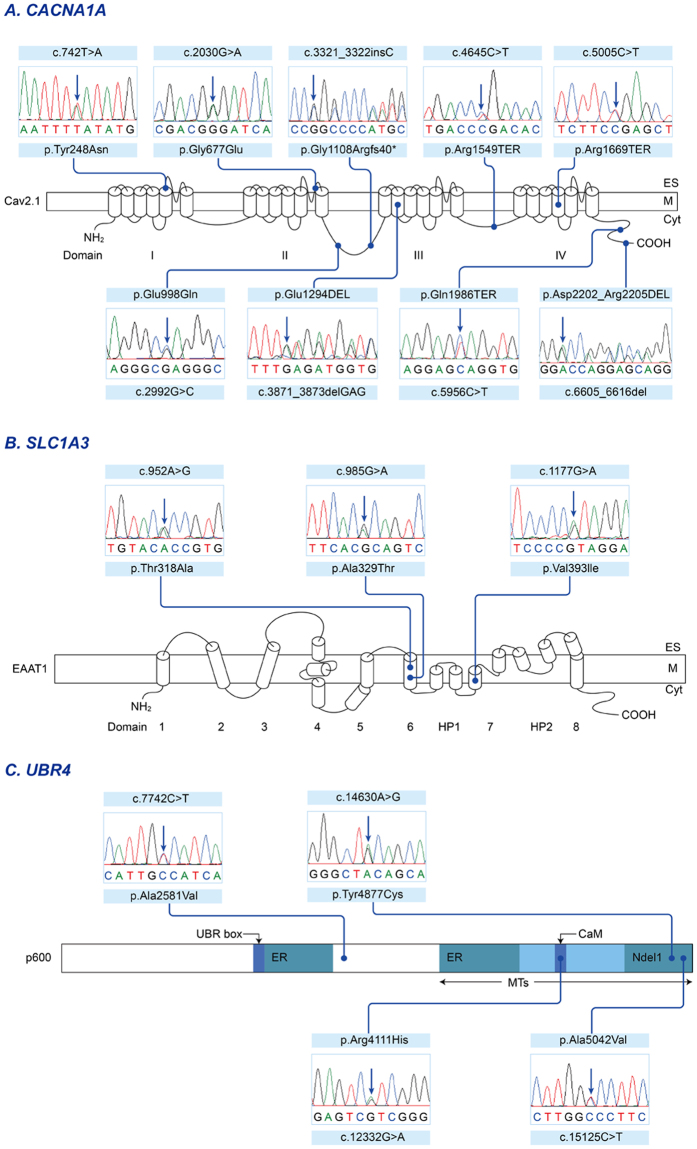
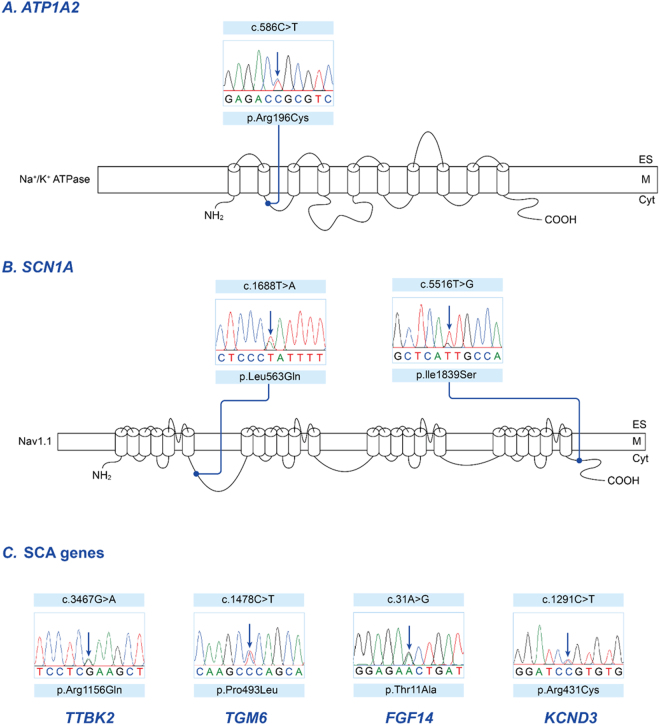
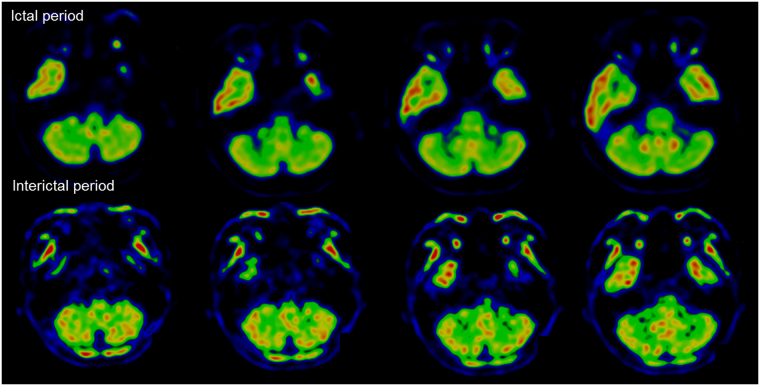
Episodic ataxia (EA) is a rare neurological condition characterized by recurrent spells of truncal ataxia and incoordination. Five genes (KCNA1, CACNA1A, CACNB4, SLC1A3, and UBR4) have been linked to EA. Despite extensive efforts to genetically diagnose EA, many patients remain still undiagnosed. Whole-exome sequencing was carried out in 39 Korean patients with EA to identify pathogenic mutations of the five known EA genes. We also evaluated 40 candidate genes that cause EA as a secondary phenotype or cerebellar ataxia. Eighteen patients (46%) revealed genetic information useful for establishing a molecular diagnosis of EA. In 11 patients, 16 pathogenic mutations were detected in three EA genes. These included nine mutations in CACNA1A, three in SLC1A3, and four in UBR4. Three patients had mutations in two genes, either CACNA1A and SLC1A3 or CACNA1A and UBR4, suggesting that SLC1A3 and UBR4 may act as genetic modifiers with synergic effects on the abnormal presynaptic activity caused by CACNA1A mutations. In seven patients with negative results for screening of EA genes, potential pathogenic mutations were identified in the candidate genes ATP1A2, SCN1A, TTBK2, TGM6, FGF14, and KCND3. This study demonstrates the genetic heterogeneity of Korean EA, and indicates that whole-exome sequencing may be useful for molecular genetic diagnosis of EA.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
