

Allosteric fine-tuning of the conformational equilibrium poises the chaperone BiP for post-translational regulation
- PMID: 29064369
- PMCID: PMC5655141
- DOI: 10.7554/eLife.29430
Allosteric fine-tuning of the conformational equilibrium poises the chaperone BiP for post-translational regulation
Abstract
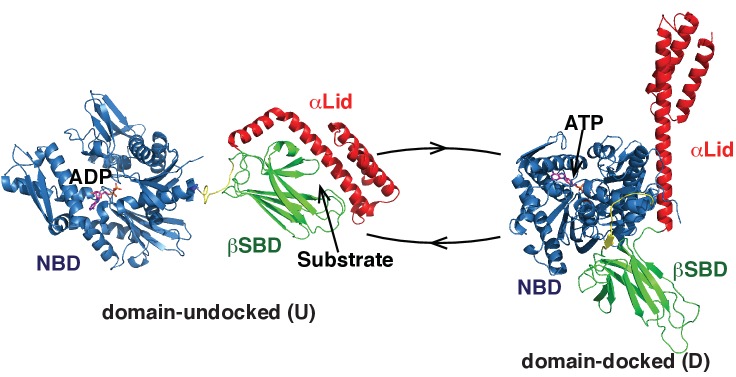
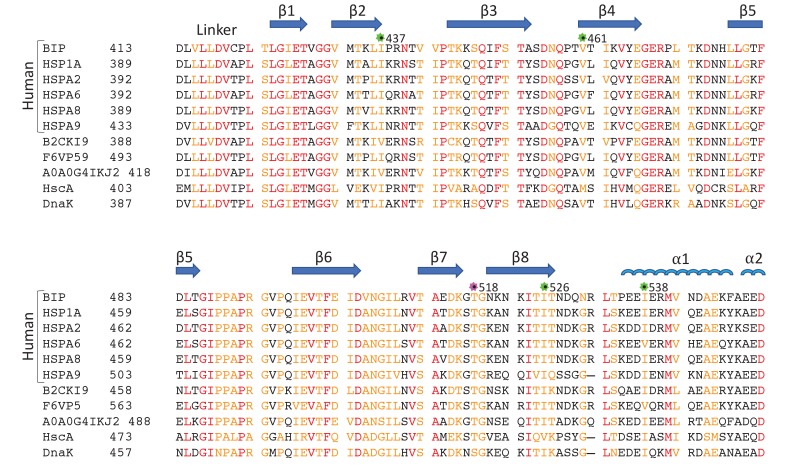
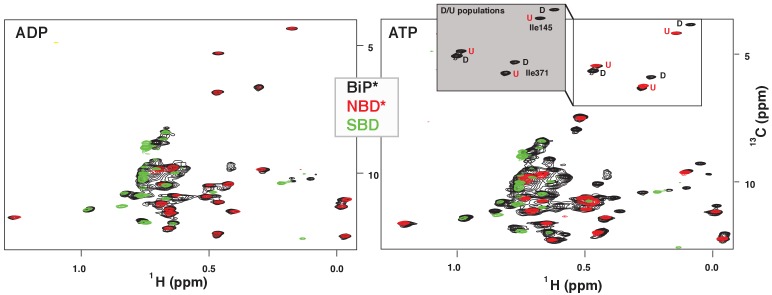
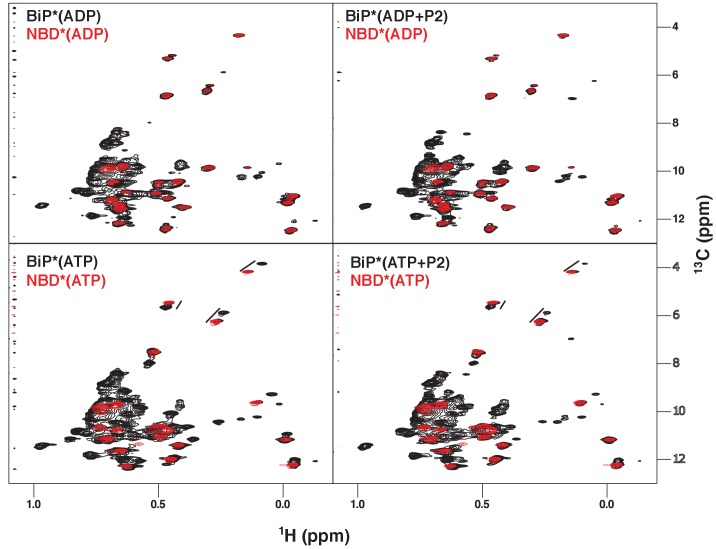
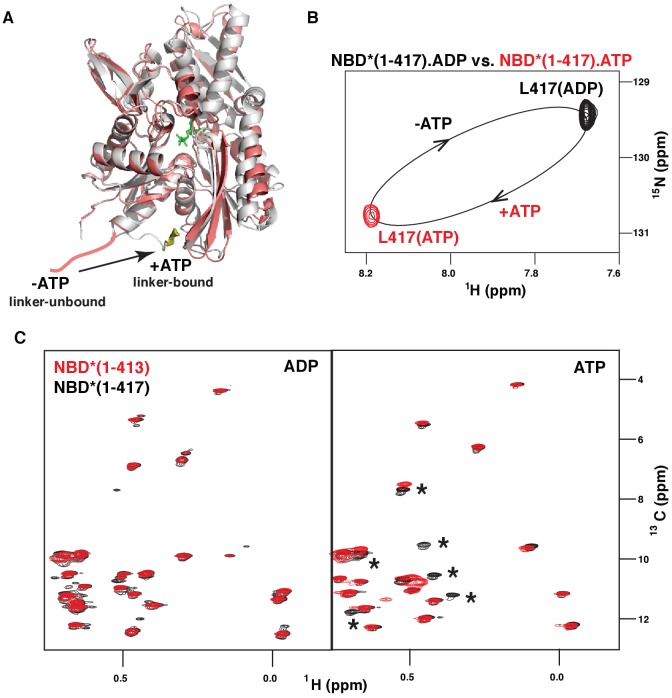
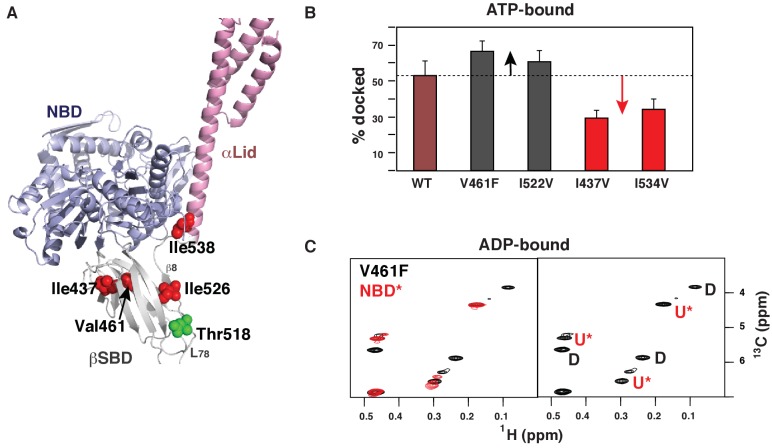
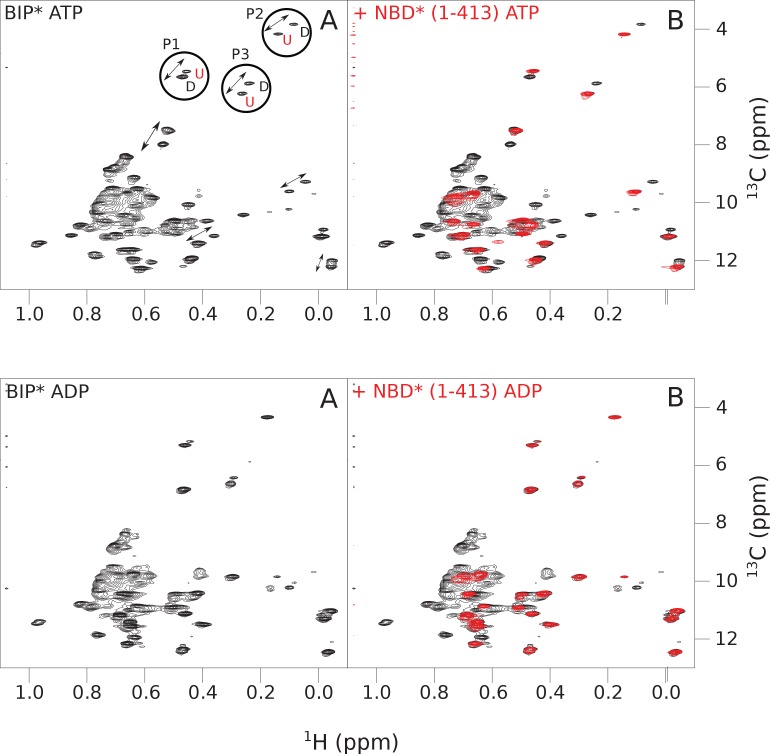
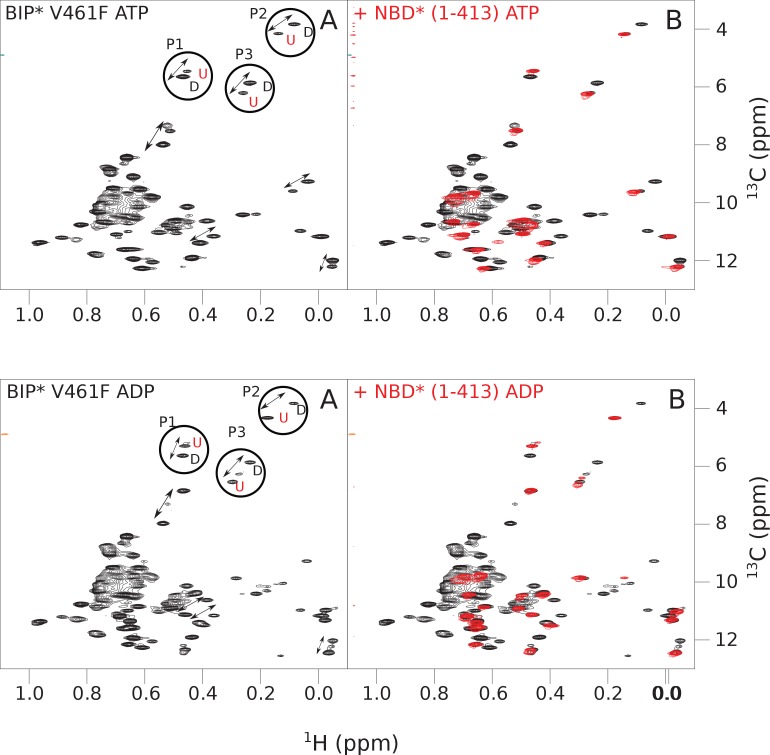
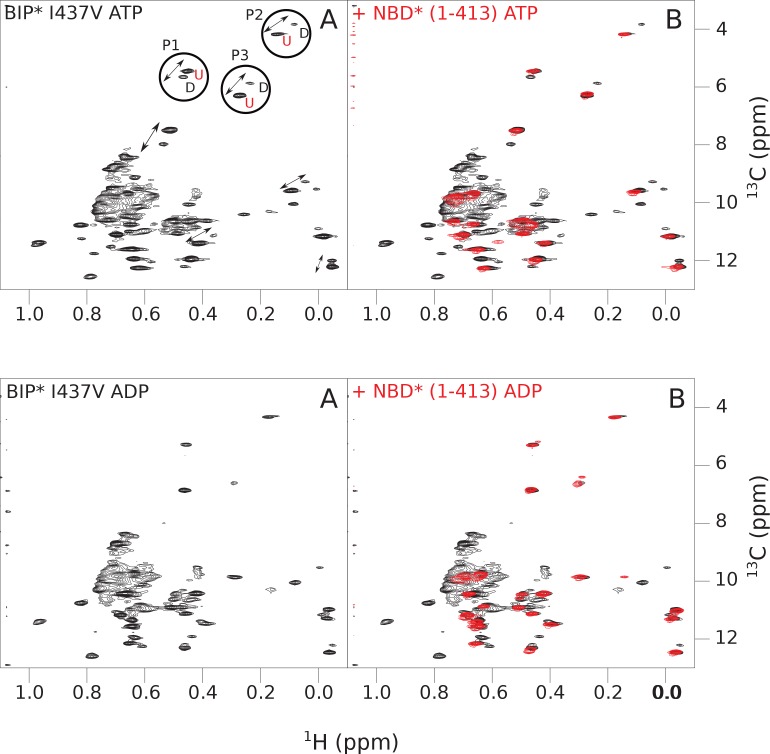
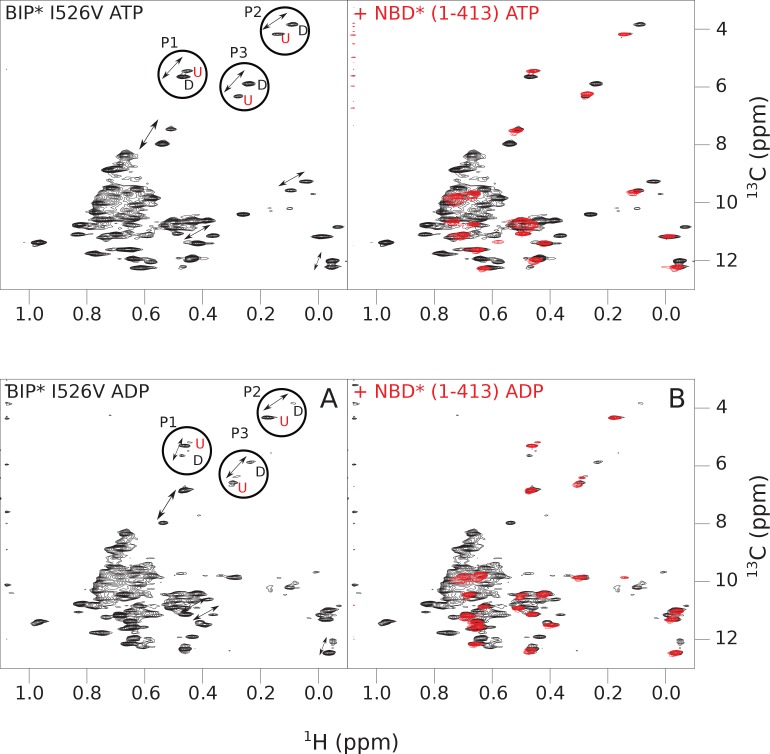
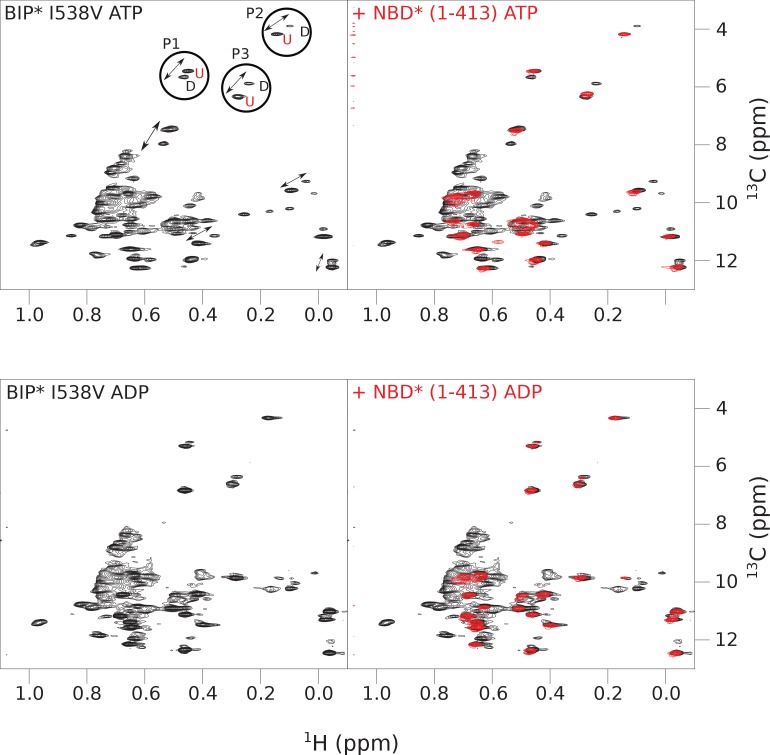
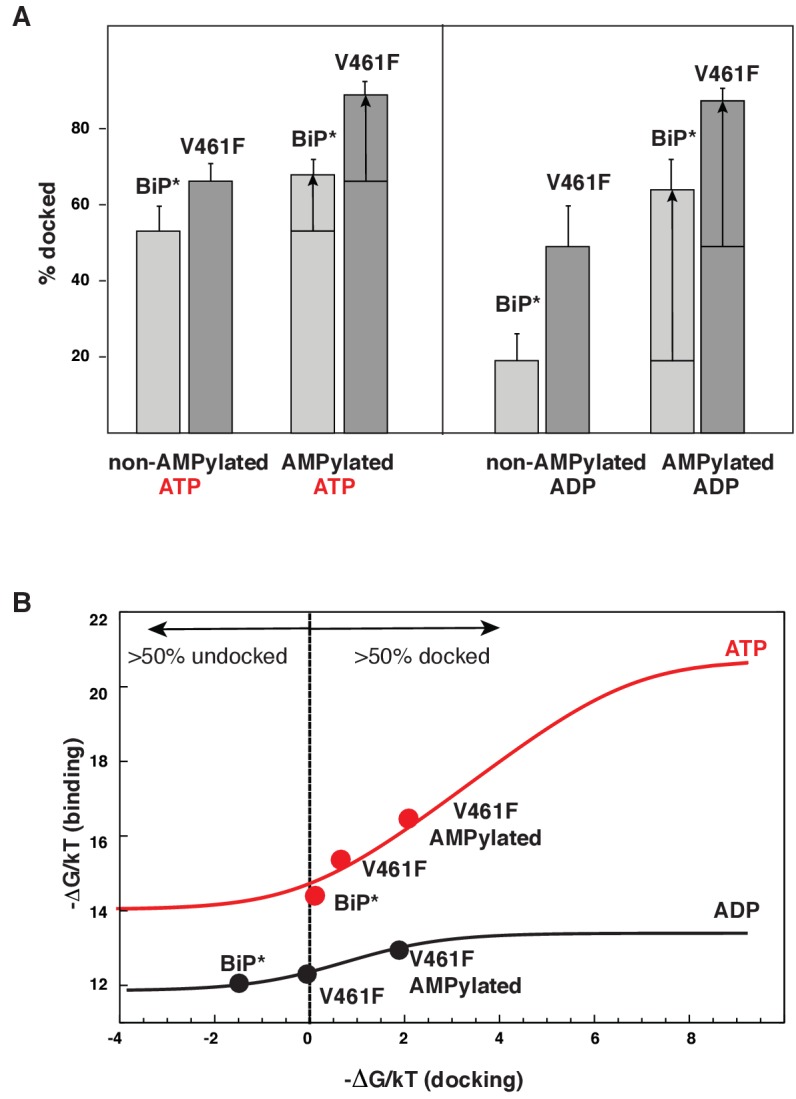
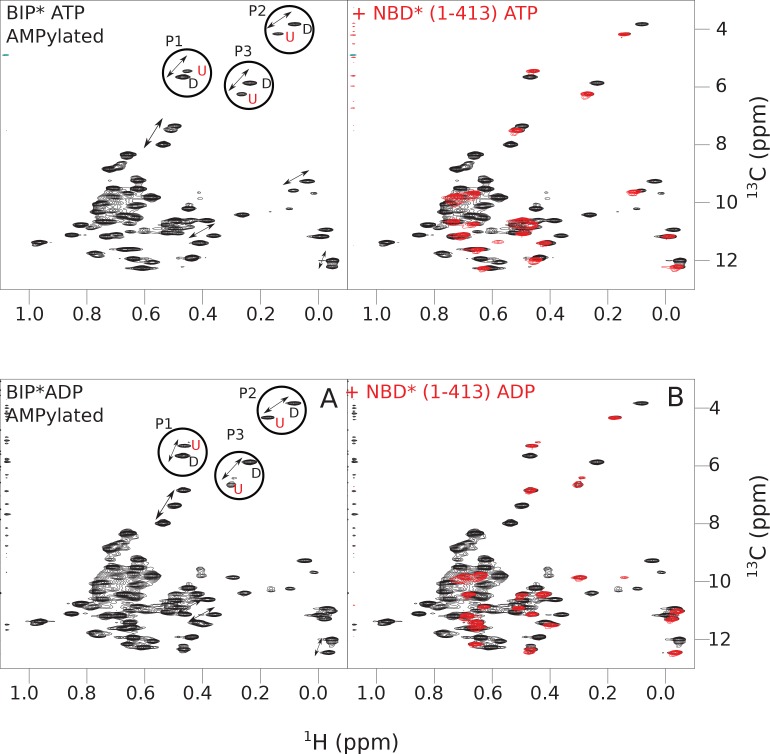
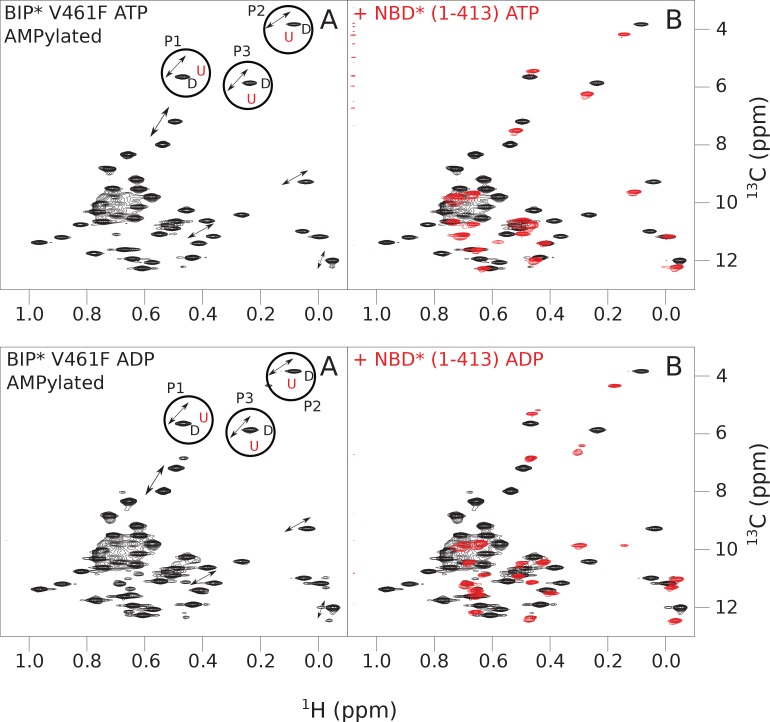
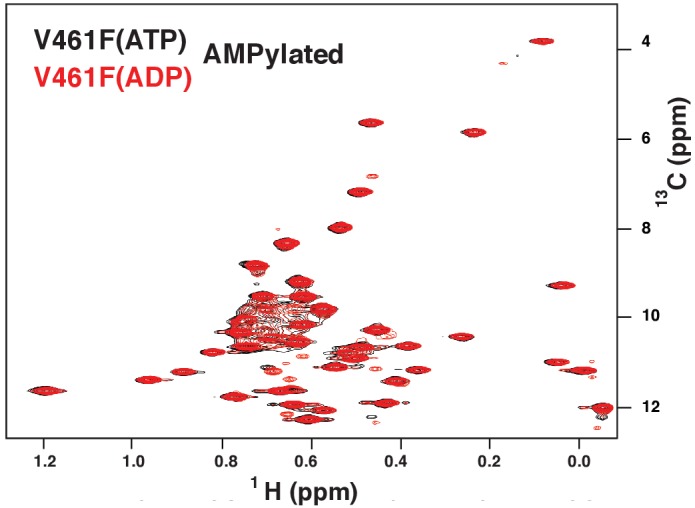
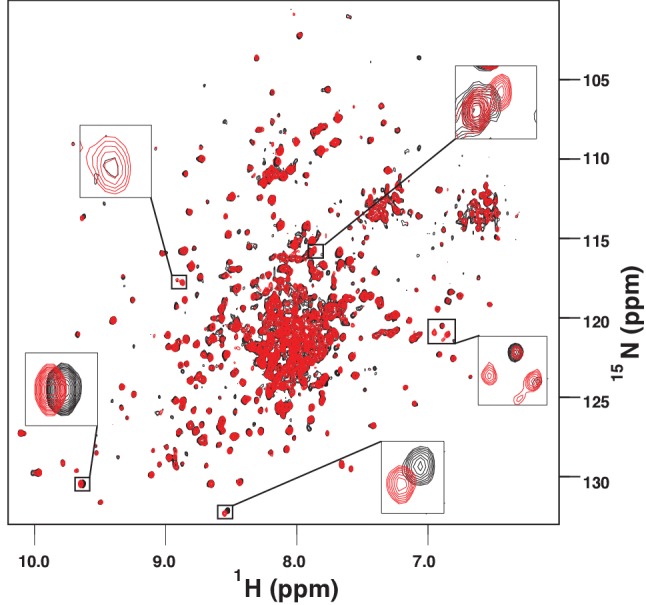
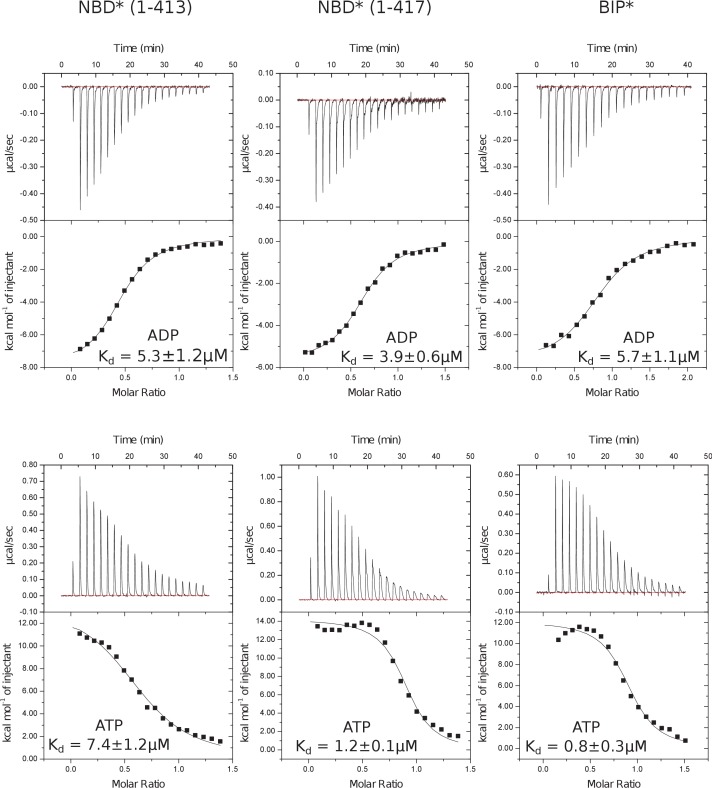
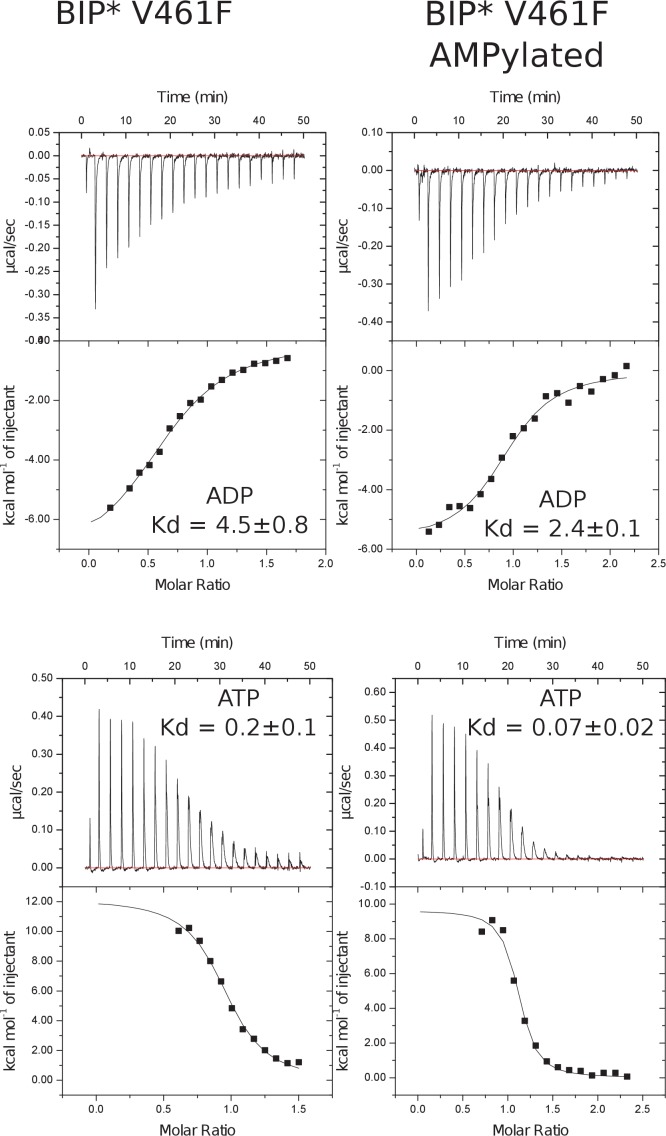
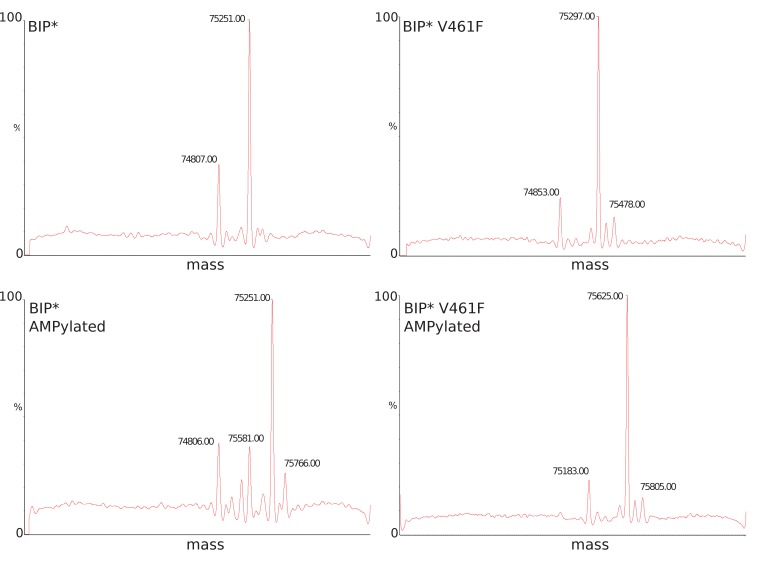
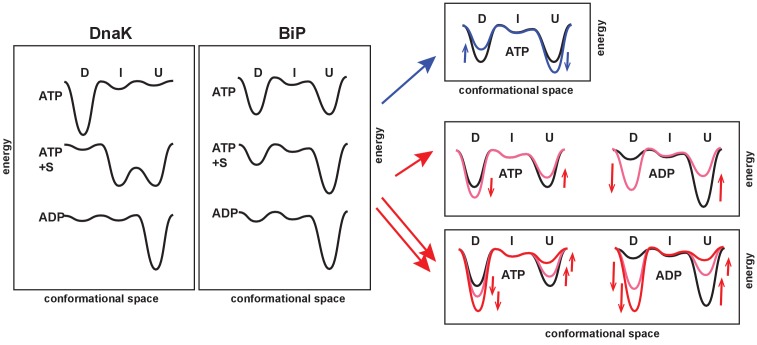
BiP is the only Hsp70 chaperone in the endoplasmic reticulum (ER) and similar to other Hsp70s, its activity relies on nucleotide- and substrate-controllable docking and undocking of its nucleotide-binding domain (NBD) and substrate-binding domain (SBD). However, little is known of specific features of the BiP conformational landscape that tune BiP to its unique tasks and the ER environment. We present methyl NMR analysis of the BiP chaperone cycle that reveals surprising conformational heterogeneity of ATP-bound BiP that distinguishes BiP from its bacterial homologue DnaK. This unusual poise enables gradual post-translational regulation of the BiP chaperone cycle and its chaperone activity by subtle local perturbations at SBD allosteric 'hotspots'. In particular, BiP inactivation by AMPylation of its SBD does not disturb Hsp70 inter-domain allostery and preserves BiP structure. Instead it relies on a redistribution of the BiP conformational ensemble and stabilization the domain-docked conformation in presence of ADP and ATP.
Keywords: AMPylation; E. coli; Hsp70; allostery; biophysics; domain docking; human; methyl NMR; structural biology.
Conflict of interest statement
No competing interests declared.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
