

MolProbity: More and better reference data for improved all-atom structure validation
- PMID: 29067766
- PMCID: PMC5734394
- DOI: 10.1002/pro.3330
MolProbity: More and better reference data for improved all-atom structure validation
Abstract
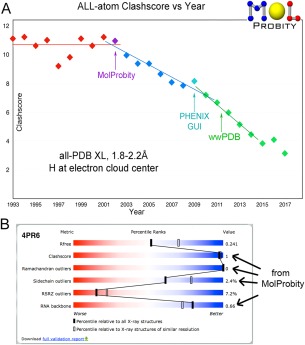
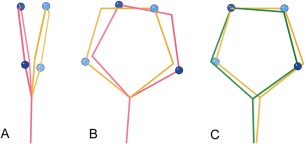
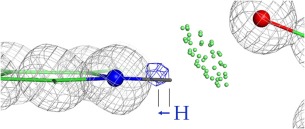
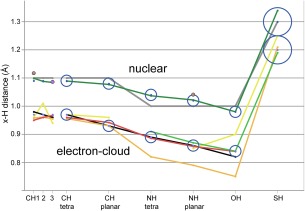
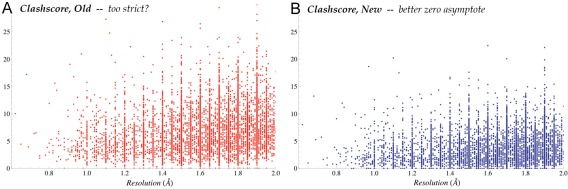
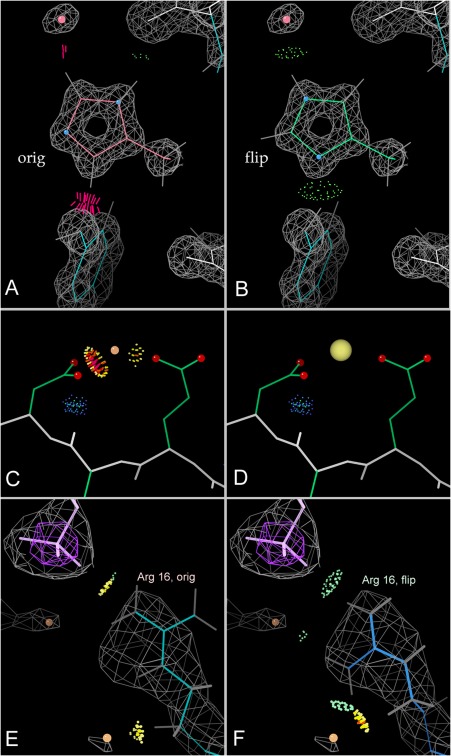
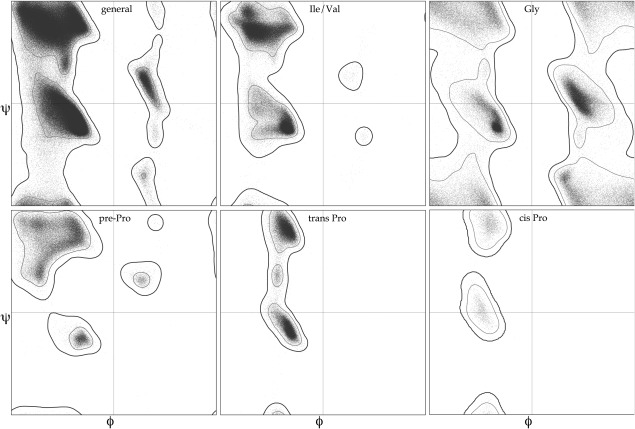
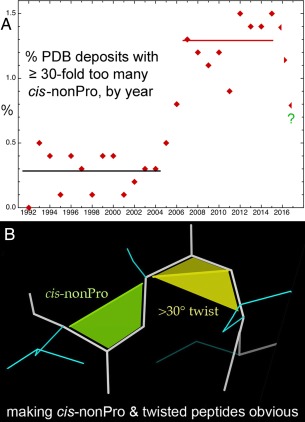
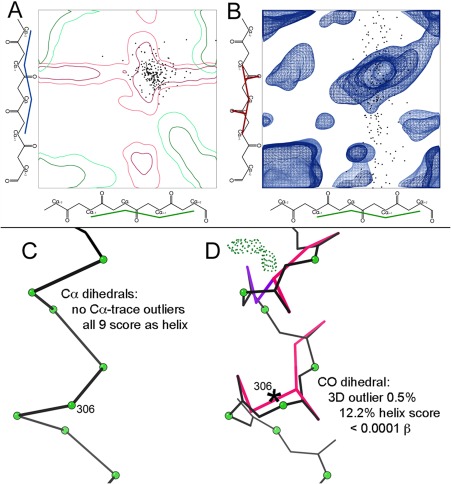
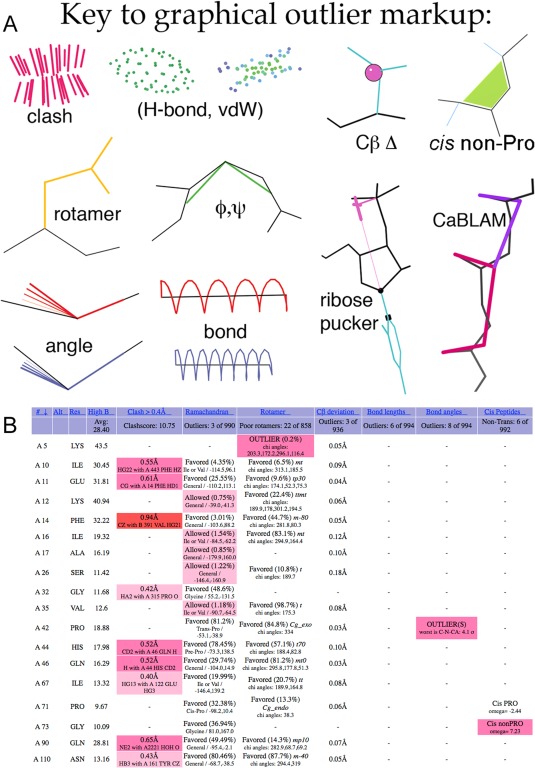
This paper describes the current update on macromolecular model validation services that are provided at the MolProbity website, emphasizing changes and additions since the previous review in 2010. There have been many infrastructure improvements, including rewrite of previous Java utilities to now use existing or newly written Python utilities in the open-source CCTBX portion of the Phenix software system. This improves long-term maintainability and enhances the thorough integration of MolProbity-style validation within Phenix. There is now a complete MolProbity mirror site at http://molprobity.manchester.ac.uk. GitHub serves our open-source code, reference datasets, and the resulting multi-dimensional distributions that define most validation criteria. Coordinate output after Asn/Gln/His "flip" correction is now more idealized, since the post-refinement step has apparently often been skipped in the past. Two distinct sets of heavy-atom-to-hydrogen distances and accompanying van der Waals radii have been researched and improved in accuracy, one for the electron-cloud-center positions suitable for X-ray crystallography and one for nuclear positions. New validations include messages at input about problem-causing format irregularities, updates of Ramachandran and rotamer criteria from the million quality-filtered residues in a new reference dataset, the CaBLAM Cα-CO virtual-angle analysis of backbone and secondary structure for cryoEM or low-resolution X-ray, and flagging of the very rare cis-nonProline and twisted peptides which have recently been greatly overused. Due to wide application of MolProbity validation and corrections by the research community, in Phenix, and at the worldwide Protein Data Bank, newly deposited structures have continued to improve greatly as measured by MolProbity's unique all-atom clashscore.
Keywords: Asn/Gln/His flip; CCTBX; CaBLAM; Top8000; all-atom contact analysis; cis non-proline; electron-cloud hydrogen position.
© 2017 The Protein Society.
Figures
References
-
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) ProCheck: a program to check the stereochemical quality of protein structures. J Appl Cryst 26:283–291.
-
- Hooft RWW, Vriend G, Sander C, Abola EE (1996) Errors in protein structures. Nature 381:272–272. - PubMed
-
- Kleywegt GJ, Jones TA (1996) Efficient rebuilding of protein structures. Acta Cryst 52:829–832. - PubMed
-
- Yeates TO (1997) Detecting and overcoming crystal twinning. Methods Enzymol 276:344–358. - PubMed
-
- Zwart PH, Grosse‐Kunstleve RW, Adams PW (2005) Xtriage and Fest: automatic assessment of X‐ray data and substructure structure factor estimation. CCP4 Newsletter, Winter, Contribution 7.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
