

Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies
- PMID: 29068549
- PMCID: PMC6198324
- DOI: 10.1002/humu.23362
Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies
Abstract
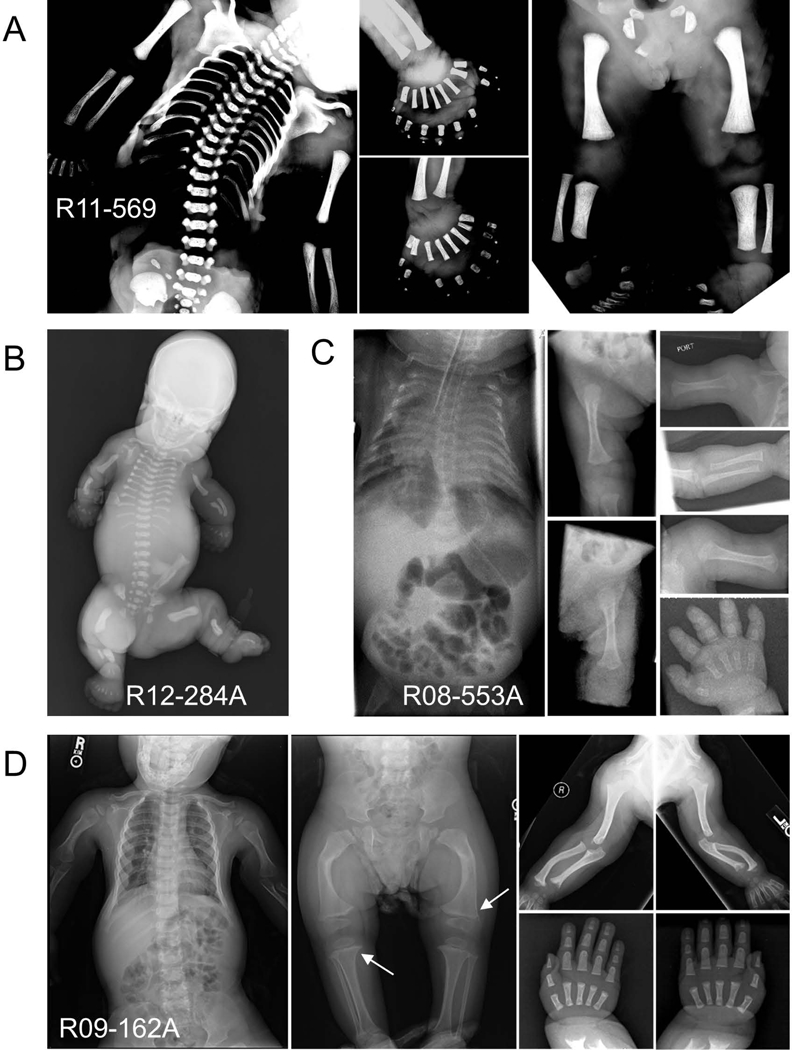
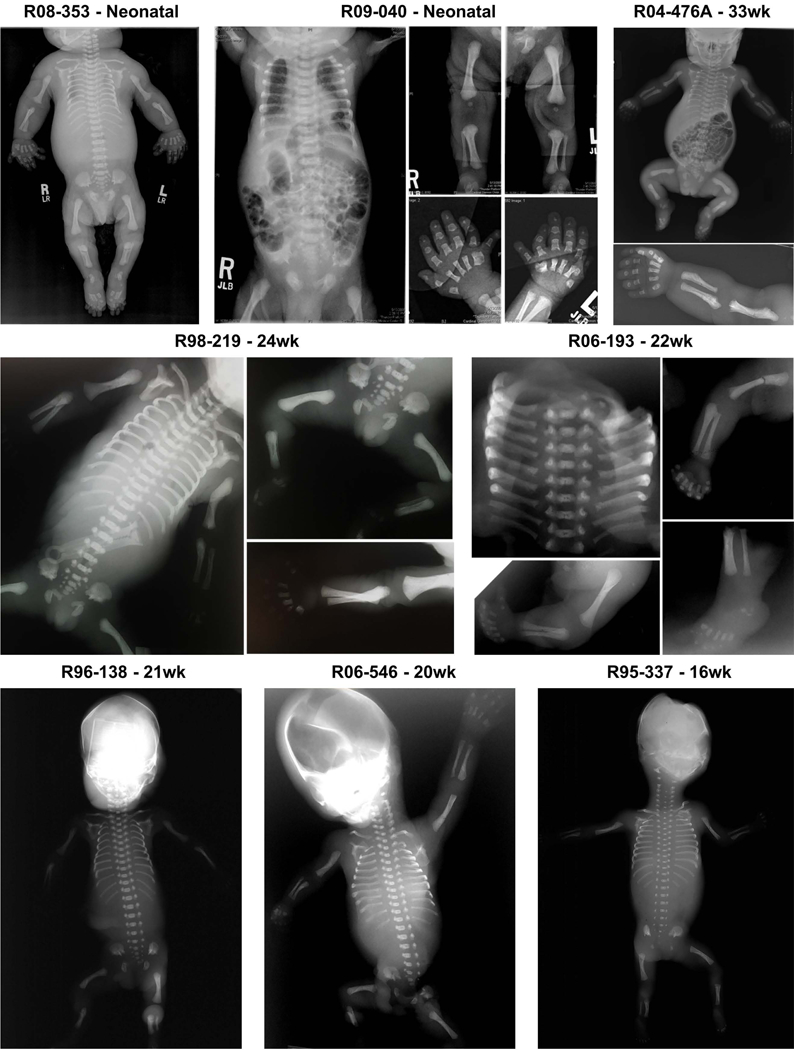
Defects in the biosynthesis and/or function of primary cilia cause a spectrum of disorders collectively referred to as ciliopathies. A subset of these disorders is distinguished by profound abnormalities of the skeleton that include a long narrow chest with markedly short ribs, extremely short limbs, and polydactyly. These include the perinatal lethal short-rib polydactyly syndromes (SRPS) and the less severe asphyxiating thoracic dystrophy (ATD), Ellis-van Creveld (EVC) syndrome, and cranioectodermal dysplasia (CED) phenotypes. To identify new genes and define the spectrum of mutations in the skeletal ciliopathies, we analyzed 152 unrelated families with SRPS, ATD, and EVC. Causal variants were discovered in 14 genes in 120 families, including one newly associated gene and two genes previously associated with other ciliopathies. These three genes encode components of three different ciliary complexes; FUZ, which encodes a planar cell polarity complex molecule; TRAF3IP1, which encodes an anterograde ciliary transport protein; and LBR, which encodes a nuclear membrane protein with sterol reductase activity. The results established the molecular basis of SRPS type IV, in which mutations were identified in four different ciliary genes. The data provide systematic insight regarding the genotypes associated with a large cohort of these genetically heterogeneous phenotypes and identified new ciliary components required for normal skeletal development.
Keywords: chondrocyte; cilia; ciliopathy; skeletal dysplasia.
© 2017 Wiley Periodicals, Inc.
Figures



References
-
- Alby C, Piquand K, Huber C, Megarbane A, Ichkou A, Legendre M, Pelluard F, Encha-Ravazi F, Abi-Tayeh G, Bessieres B, El Chehadeh-Djebbar S, Laurent N, Faivre L, Sztriha L, Zombor M, Szabo H, Failler M, Garfa-Traore M, Bole C, Nitschke P, Nizon M, Elkhartoufi N, Clerget-Darpoux F, Munnich A, Lyonnet S, Vekemans M, Saunier S, Cormier-Daire V, Attie-Bitach T, Thomas S. 2015. Mutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome. American Journal of Human Genetics 97(2):311–318. - PMC - PubMed
-
- Arts HH, Bongers EMHF, Mans DA, van Beersum SEC, Oud MM, Bolat E, Spruijt L, Cornelissen EAM, Schuurs-Hoeijmakers JHM, de Leeuw N, Cormier-Daire V, Brunner HG, Knoers NVAM, Roepman R. 2011. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. Journal of Medical Genetics 48(6):390–395. - PubMed
-
- Badano JL, Mitsuma N, Beales PL, Katsanis N. 2006. The ciliopathies: An emerging class of human genetic disorders. Annual Review of Genomics and Human Genetics 7:125–148. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
