

Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells
- PMID: 29069777
- PMCID: PMC5641120
- DOI: 10.18632/oncotarget.20050
Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells
Abstract
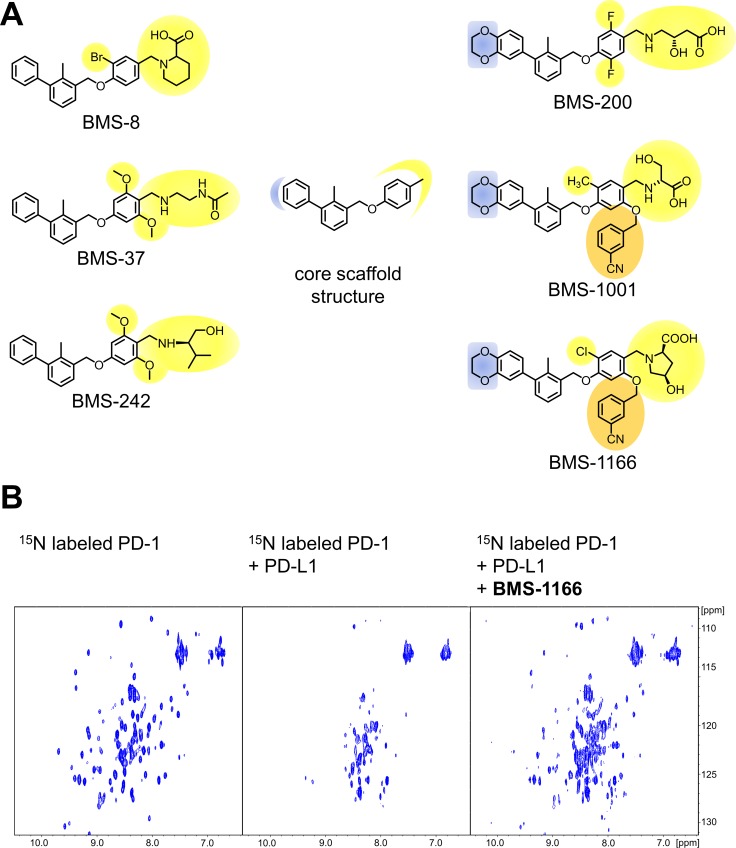
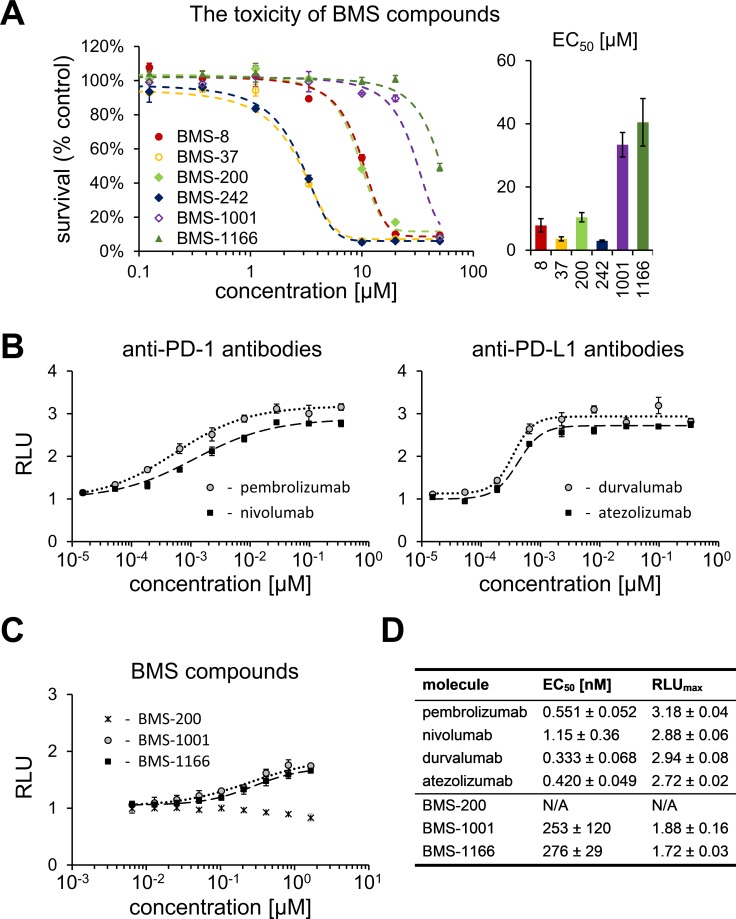
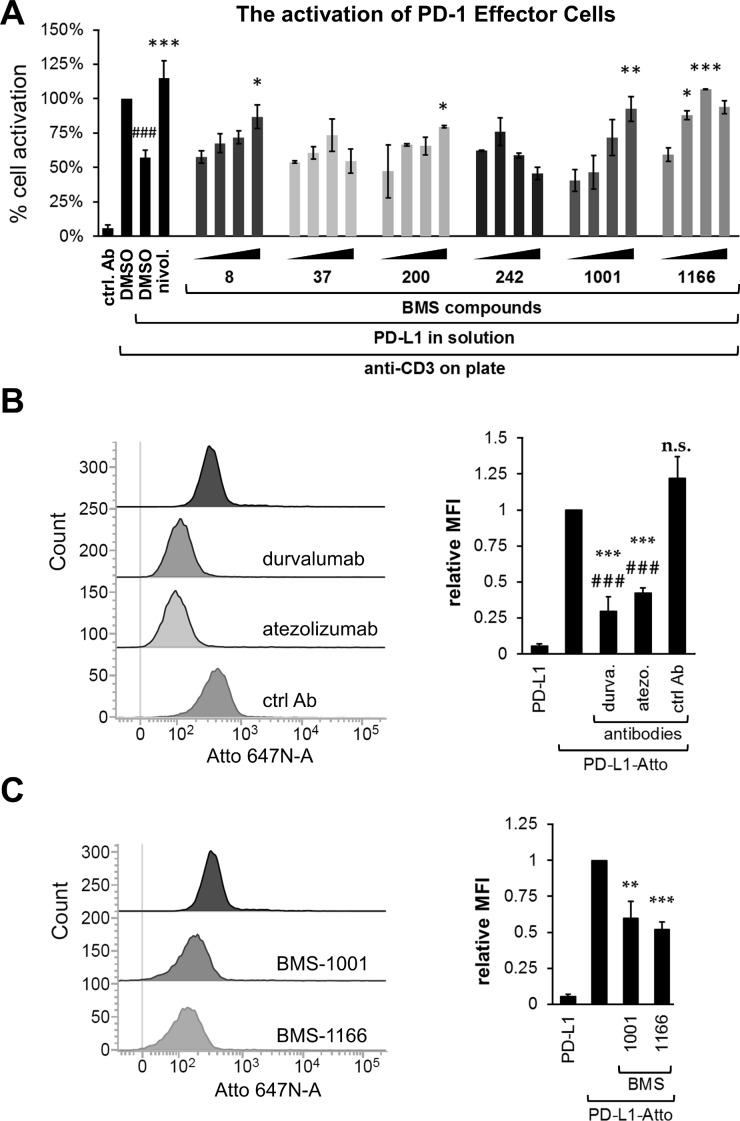
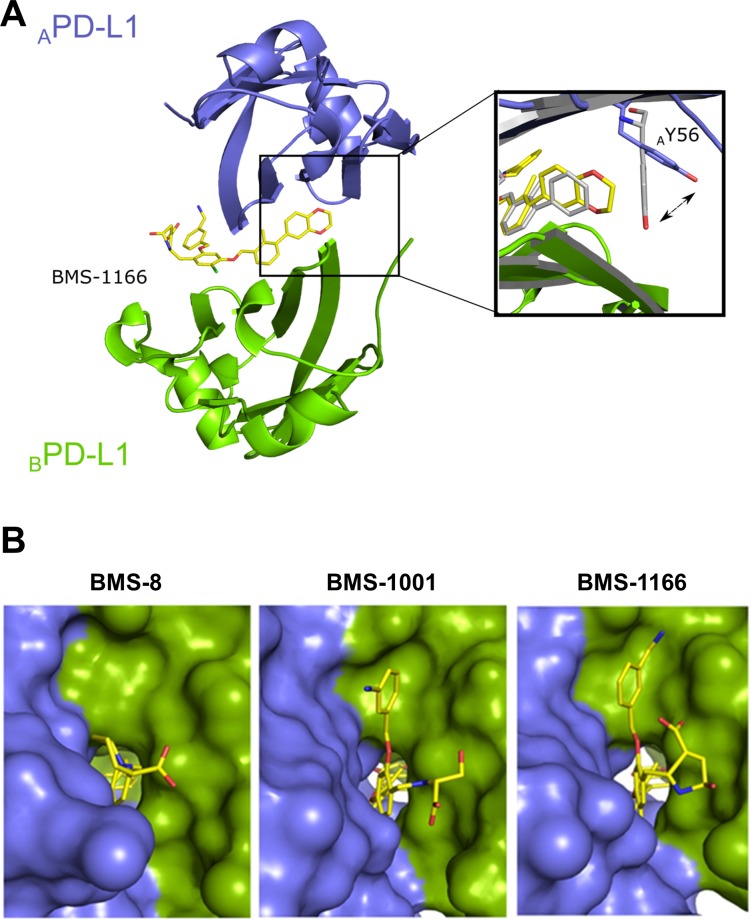
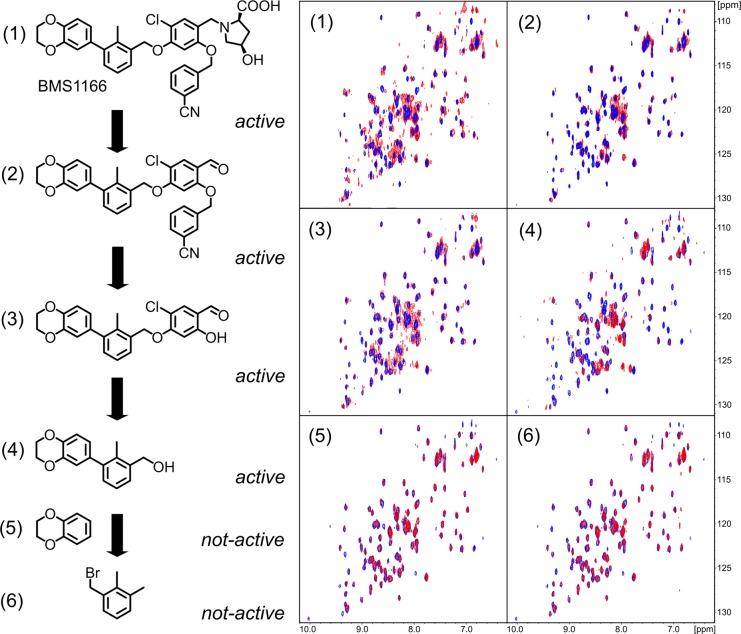
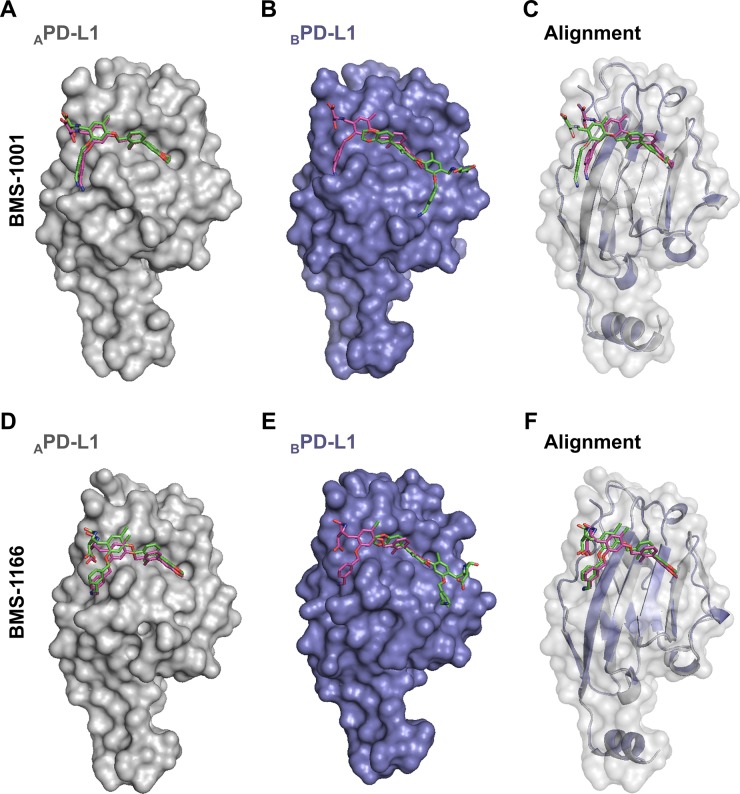
Antibodies targeting the PD-1/PD-L1 immune checkpoint achieved spectacular success in anticancer therapy in the recent years. In contrast, no small molecules with cellular activity have been reported so far. Here we provide evidence that small molecules are capable of alleviating the PD-1/PD-L1 immune checkpoint-mediated exhaustion of Jurkat T-lymphocytes. The two optimized small-molecule inhibitors of the PD-1/PD-L1 interaction, BMS-1001 and BMS-1166, developed by Bristol-Myers Squibb, bind to human PD-L1 and block its interaction with PD-1, when tested on isolated proteins. The compounds present low toxicity towards tested cell lines and block the interaction of soluble PD-L1 with the cell surface-expressed PD-1. As a result, BMS-1001 and BMS-1166 alleviate the inhibitory effect of the soluble PD-L1 on the T-cell receptor-mediated activation of T-lymphocytes. Moreover, the compounds were effective in attenuating the inhibitory effect of the cell surface-associated PD-L1. We also determined the X-ray structures of the complexes of BMS-1001 and BMS-1166 with PD-L1, which revealed features that may be responsible for increased potency of the compounds compared to their predecessors. Further development may lead to the design of an anticancer therapy based on the orally delivered immune checkpoint inhibition.
Keywords: PD-1; PD-L1; immune checkpoint blockade; inhibitor; small-molecules.
Conflict of interest statement
CONFLICTS OF INTEREST The authors declare no conflicts of interests.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
