

The E3 ubiquitin ligase SCFFBXL14 complex stimulates neuronal differentiation by targeting the Notch signaling factor HES1 for proteolysis
- PMID: 29070679
- PMCID: PMC5723999
- DOI: 10.1074/jbc.M117.815001
The E3 ubiquitin ligase SCFFBXL14 complex stimulates neuronal differentiation by targeting the Notch signaling factor HES1 for proteolysis
Abstract
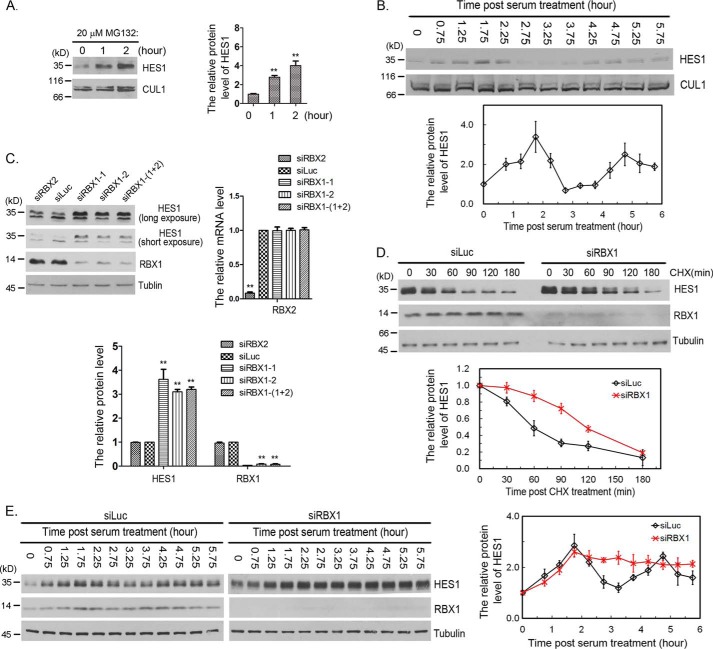
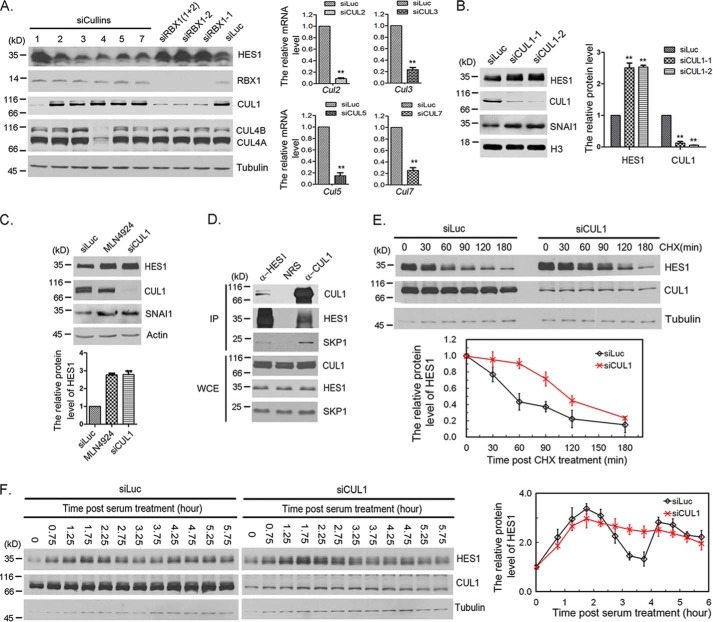
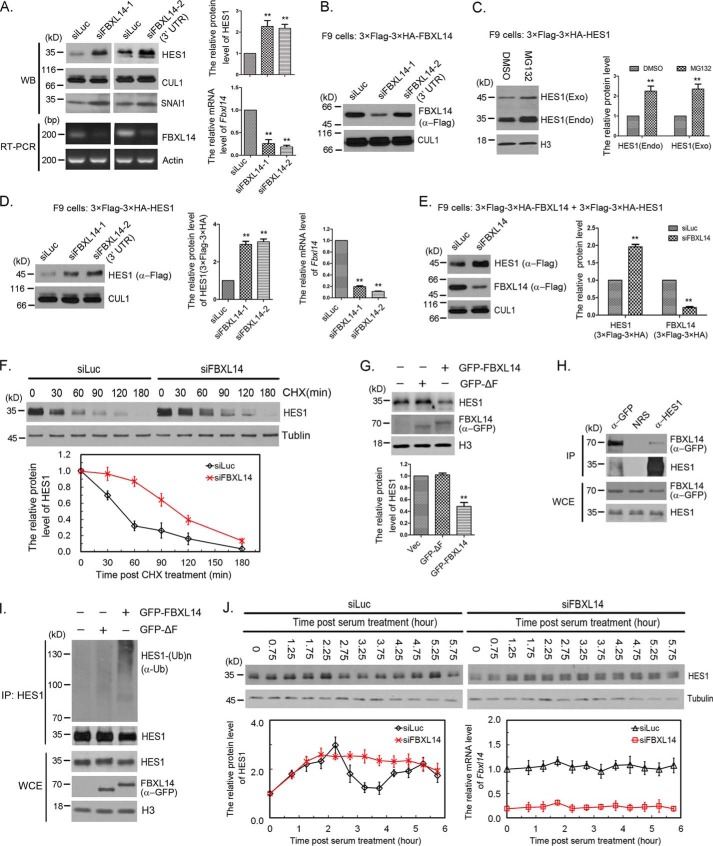
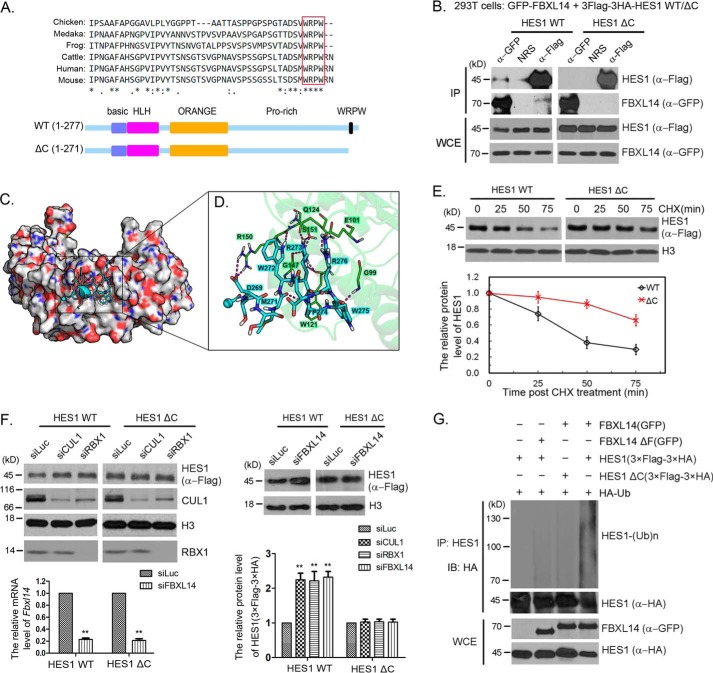
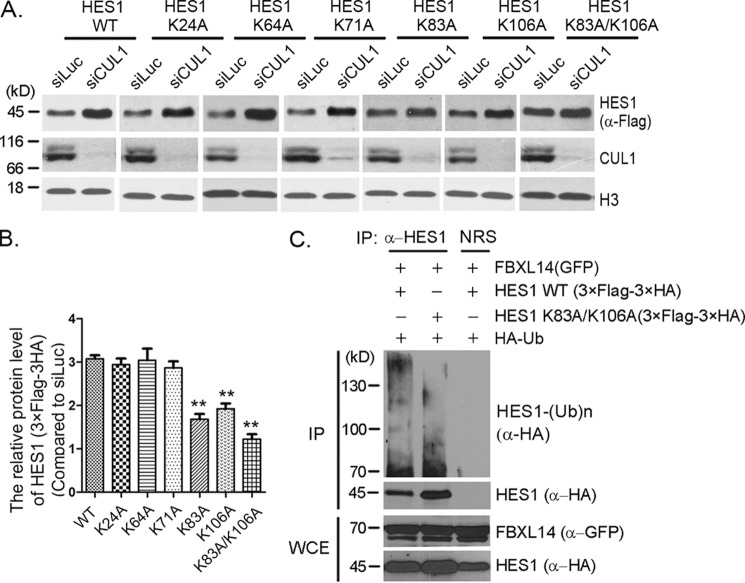
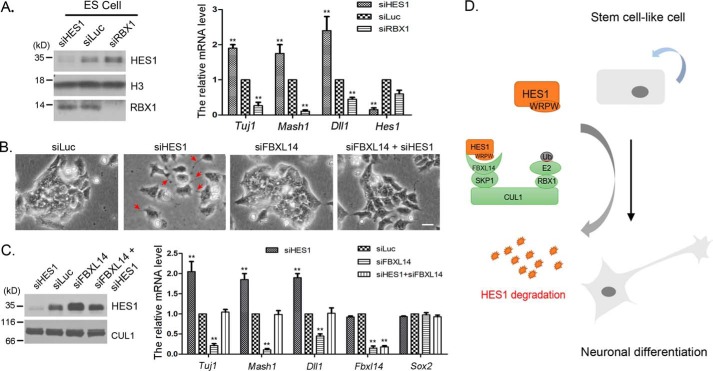
Molecular oscillators are important cellular regulators of, for example, circadian clocks, oscillations of immune regulators, and short-period (ultradian) rhythms during embryonic development. The Notch signaling factor HES1 (hairy and enhancer of split 1) is a well-known repressor of proneural genes, and HES1 ultradian oscillation is essential for keeping cells in an efficiently proliferating progenitor state. HES1 oscillation is driven by both transcriptional self-repression and ubiquitin-dependent proteolysis. However, the E3 ubiquitin ligase targeting HES1 for proteolysis remains unclear. Based on siRNA-mediated gene silencing screening, co-immunoprecipitation, and ubiquitination assays, we discovered that the E3 ubiquitin ligase SCFFBXL14 complex regulates HES1 ubiquitination and proteolysis. siRNA-mediated knockdown of the Cullin-RING E3 ubiquitin ligases RBX1 or CUL1 increased HES1 protein levels, prolonged its half-life, and dampened its oscillation. FBXL14, an F-box protein for SCF ubiquitin ligase, associates with HES1. FBXL14 silencing stabilized HES1, whereas FBXL14 overexpression decreased HES1 protein levels. Of note, the SCFFBXL14 complex promoted the ubiquitination of HES1 in vivo, and a conserved WRPW motif in HES1 was essential for HES1 binding to FBXL14 and for ubiquitin-dependent HES1 degradation. HES1 knockdown promoted neuronal differentiation, but FBXL14 silencing inhibited neuronal differentiation induced by HES1 ablation in mES and F9 cells. Our results suggest that SCFFBXL14 promotes neuronal differentiation by targeting HES1 for ubiquitin-dependent proteolysis and that the C-terminal WRPW motif in HES1 is required for this process.
Keywords: E3 ubiquitin ligase; FBXL14; HES1 oscillation; SCF ubiquitin ligase; neurodifferentiation; proteolysis; small interfering RNA (siRNA); ubiquitylation (ubiquitination).
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Dunlap J. C. (1999) Molecular bases for circadian clocks. Cell 96, 271–290 - PubMed
-
- Schibler U., and Sassone-Corsi P. (2002) A web of circadian pacemakers. Cell 111, 919–922 - PubMed
-
- Tyson J. J., Chen K., and Novak B. (2001) Network dynamics and cell physiology. Nat. Rev. Mol. Cell Biol. 2, 908–916 - PubMed
-
- Pourquié O. (2003) The segmentation clock: converting embryonic time into spatial pattern. Science 301, 328–330 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
