

Small-molecule inhibitors directly target CARD9 and mimic its protective variant in inflammatory bowel disease
- PMID: 29073062
- PMCID: PMC5664502
- DOI: 10.1073/pnas.1705748114
Small-molecule inhibitors directly target CARD9 and mimic its protective variant in inflammatory bowel disease
Abstract
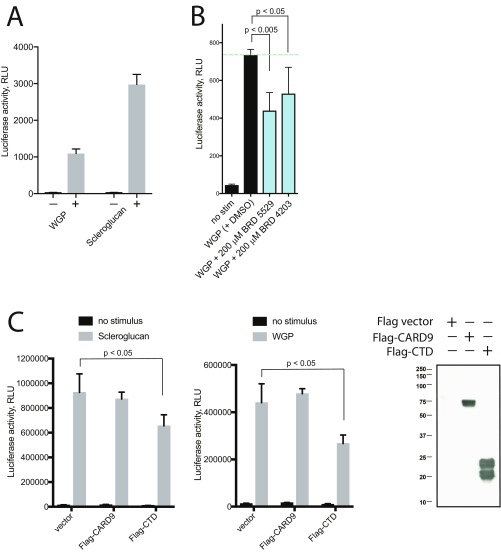
Advances in human genetics have dramatically expanded our understanding of complex heritable diseases. Genome-wide association studies have identified an allelic series of CARD9 variants associated with increased risk of or protection from inflammatory bowel disease (IBD). The predisposing variant of CARD9 is associated with increased NF-κB-mediated cytokine production. Conversely, the protective variant lacks a functional C-terminal domain and is unable to recruit the E3 ubiquitin ligase TRIM62. Here, we used biochemical insights into CARD9 variant proteins to create a blueprint for IBD therapeutics and recapitulated the mechanism of the CARD9 protective variant using small molecules. We developed a multiplexed bead-based technology to screen compounds for disruption of the CARD9-TRIM62 interaction. We identified compounds that directly and selectively bind CARD9, disrupt TRIM62 recruitment, inhibit TRIM62-mediated ubiquitinylation of CARD9, and demonstrate cellular activity and selectivity in CARD9-dependent pathways. Taken together, small molecules targeting CARD9 illustrate a path toward improved IBD therapeutics.
Keywords: CARD9; inflammatory bowel disease; small molecules; therapeutics.
Published under the PNAS license.
Conflict of interest statement
Conflict of interest statement: S.L.S. and B.F.C. were coauthors on a 2015 review article. S.L.S. and K.M.S. were coauthors on a 2015 research paper. B.F.C. and K.M.S. independently supplied reagents to the same laboratory. There was no active collaboration.
Figures
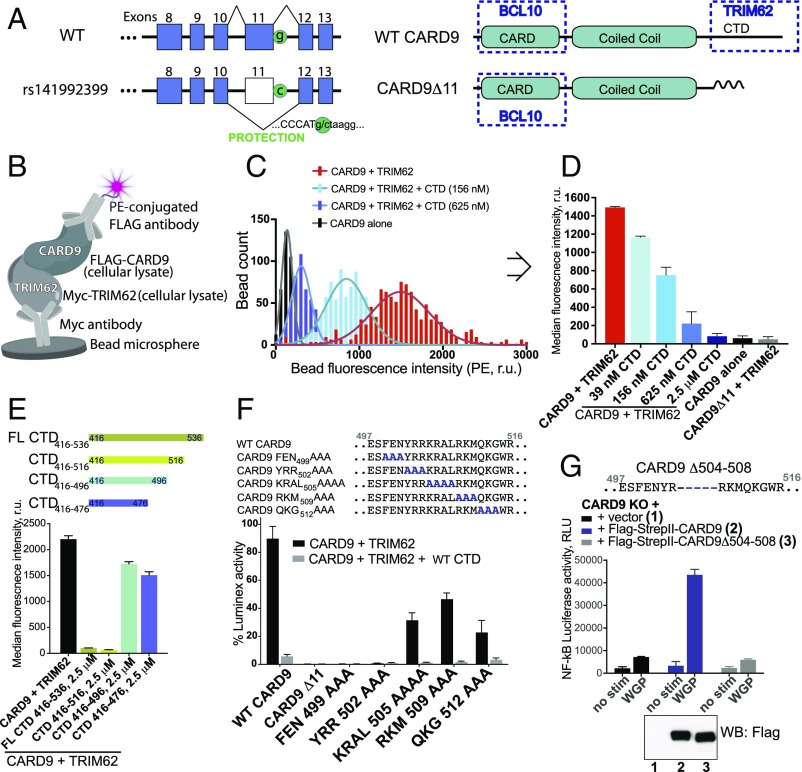
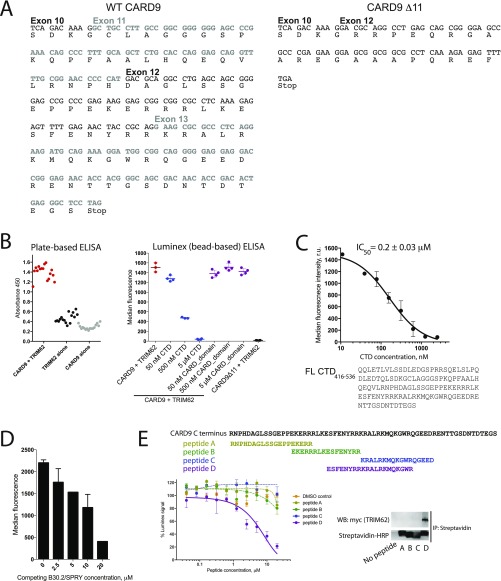
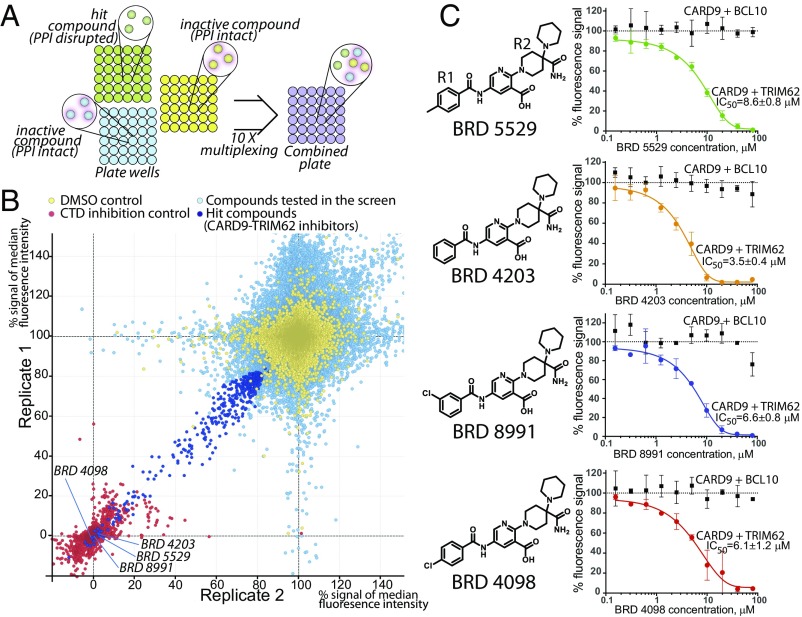
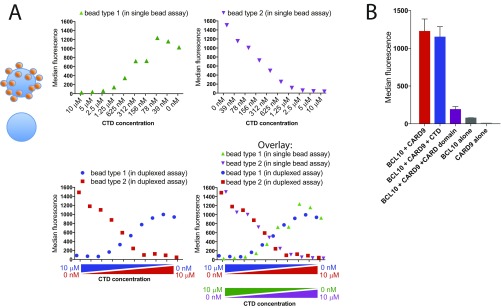
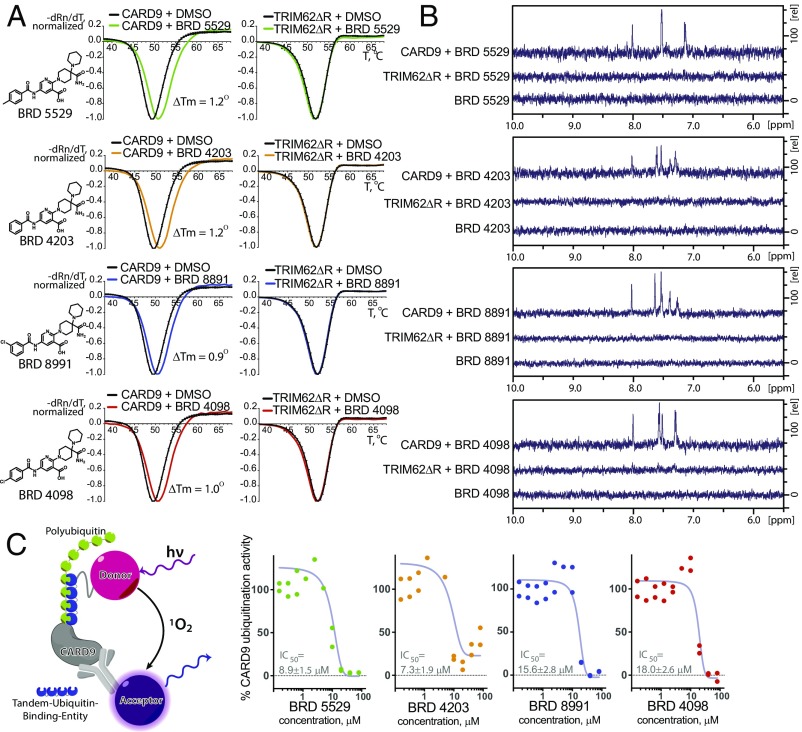
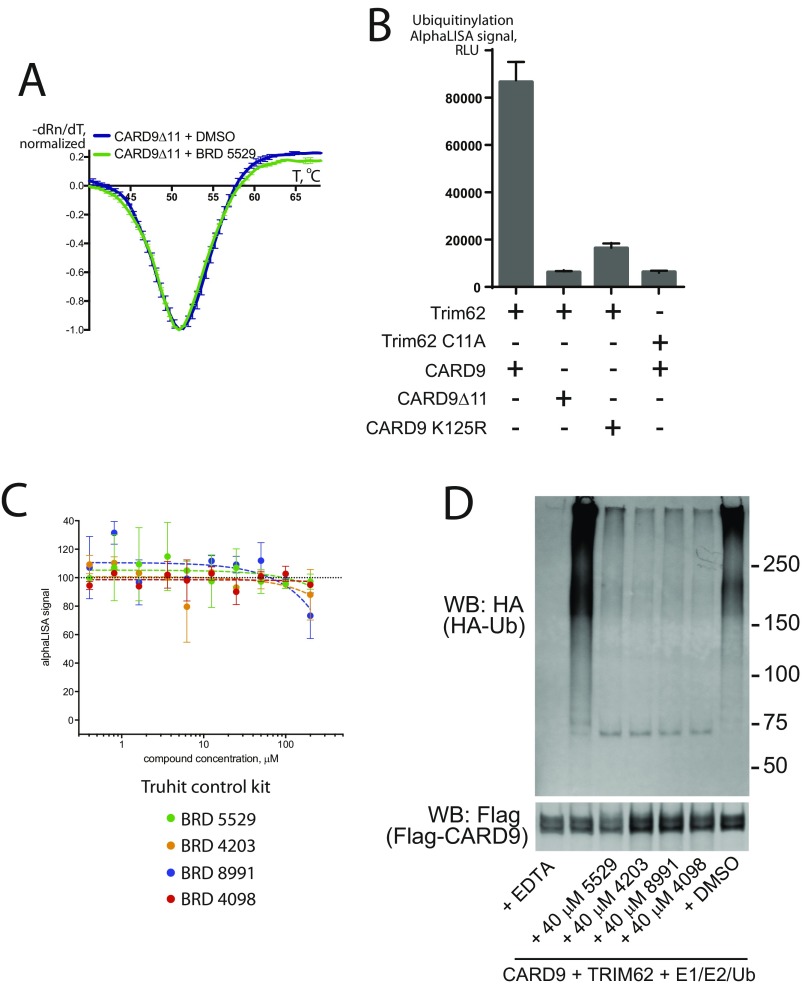
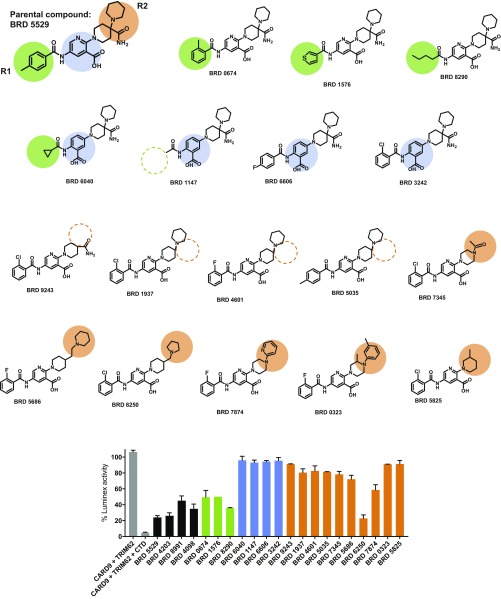
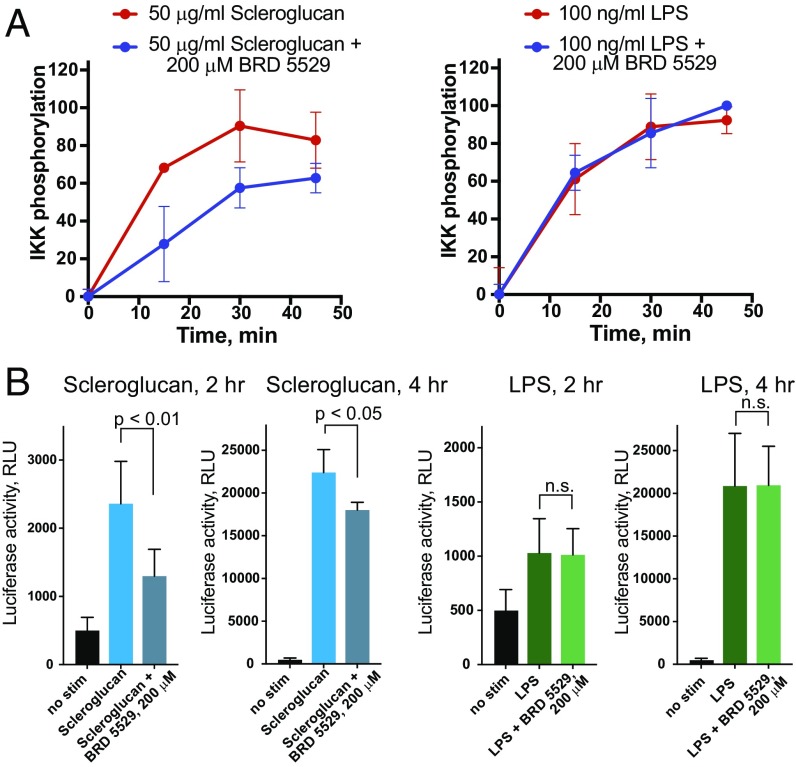
References
-
- Rivas MA, et al. National Institute of Diabetes and Digestive Kidney Diseases Inflammatory Bowel Disease Genetics Consortium (NIDDK IBDGC); United Kingdom Inflammatory Bowel Disease Genetics Consortium; International Inflammatory Bowel Disease Genetics Consortium Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
