

Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer
- PMID: 29073103
- PMCID: PMC5664494
- DOI: 10.1073/pnas.1704862114
Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer
Erratum in
-
Correction for Sansone et al., Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer.Proc Natl Acad Sci U S A. 2017 Nov 21;114(47):E10255. doi: 10.1073/pnas.1718630114. Epub 2017 Nov 13. Proc Natl Acad Sci U S A. 2017. PMID: 29133404 Free PMC article. No abstract available.
Abstract
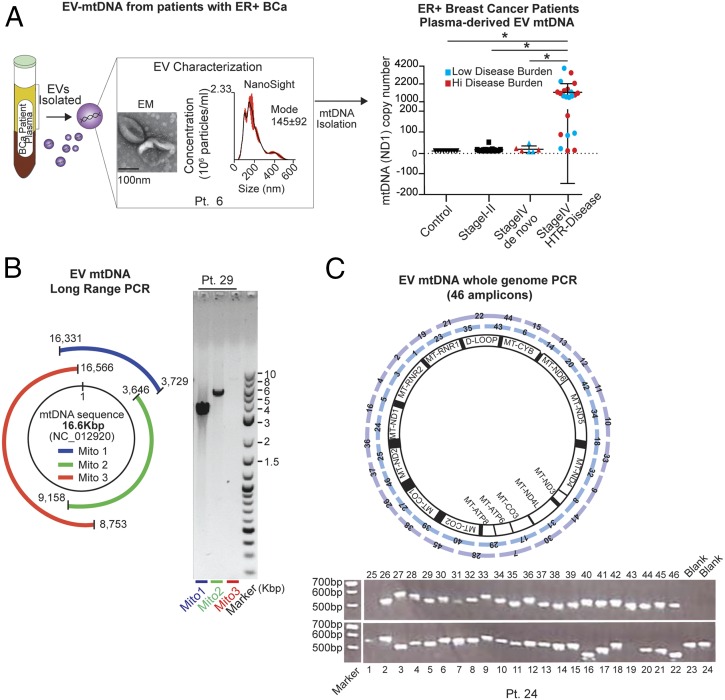
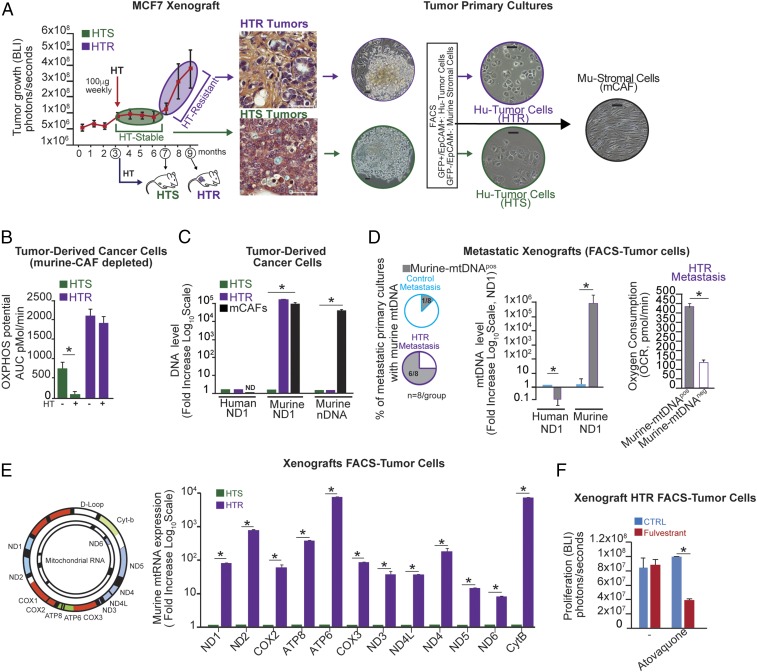
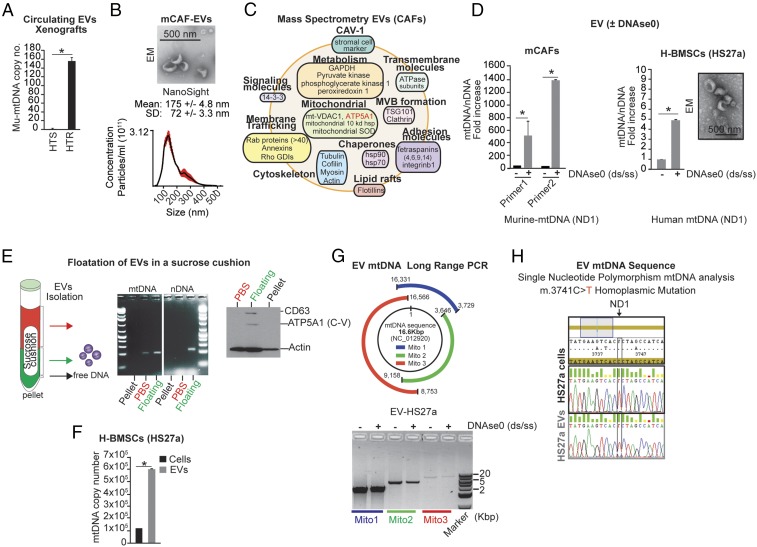
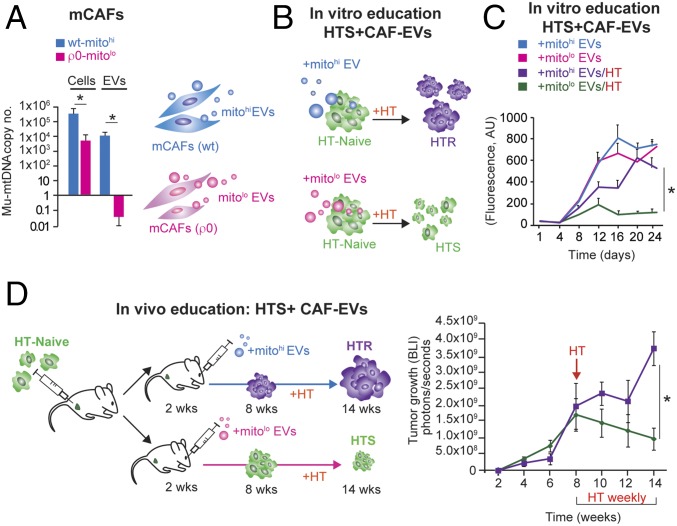
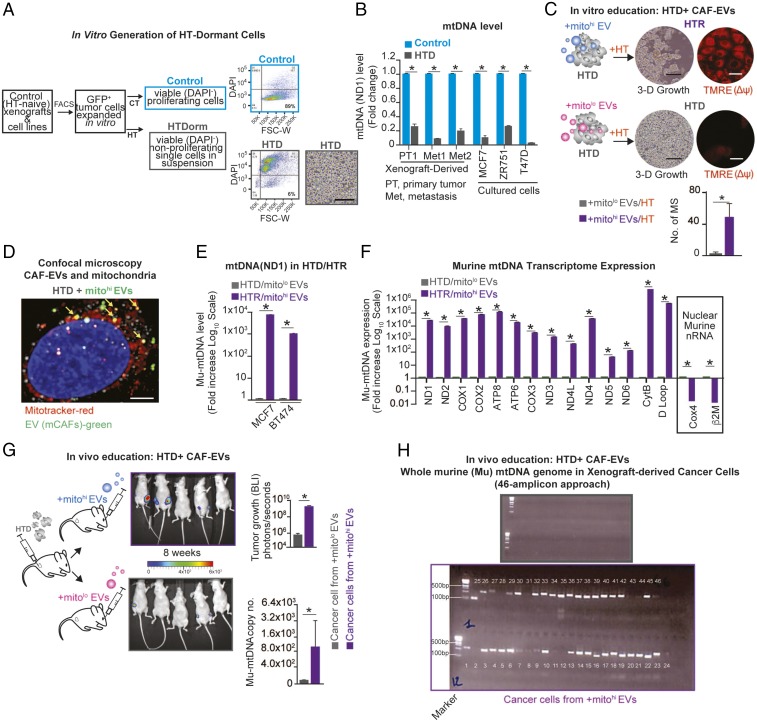
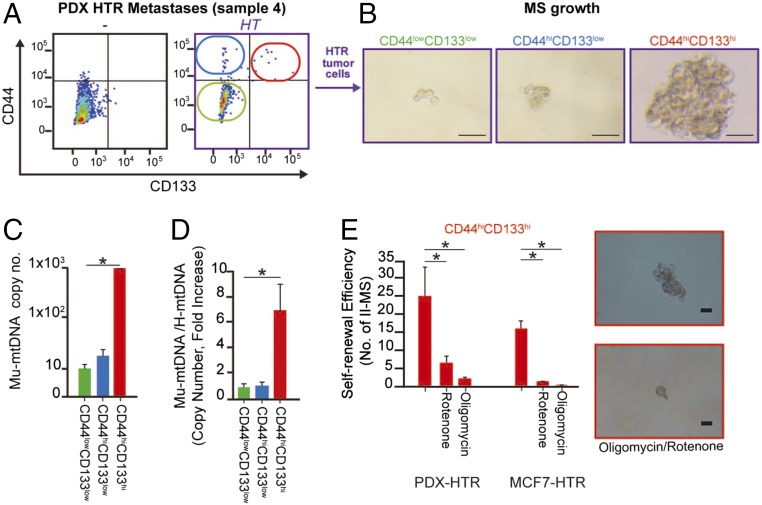
The horizontal transfer of mtDNA and its role in mediating resistance to therapy and an exit from dormancy have never been investigated. Here we identified the full mitochondrial genome in circulating extracellular vesicles (EVs) from patients with hormonal therapy-resistant (HTR) metastatic breast cancer. We generated xenograft models of HTR metastatic disease characterized by EVs in the peripheral circulation containing mtDNA. Moreover, these human HTR cells had acquired host-derived (murine) mtDNA promoting estrogen receptor-independent oxidative phosphorylation (OXPHOS). Functional studies identified cancer-associated fibroblast (CAF)-derived EVs (from patients and xenograft models) laden with whole genomic mtDNA as a mediator of this phenotype. Specifically, the treatment of hormone therapy (HT)-naive cells or HT-treated metabolically dormant populations with CAF-derived mtDNAhi EVs promoted an escape from metabolic quiescence and HTR disease both in vitro and in vivo. Moreover, this phenotype was associated with the acquisition of EV mtDNA, especially in cancer stem-like cells, expression of EV mtRNA, and restoration of OXPHOS. In summary, we have demonstrated that the horizontal transfer of mtDNA from EVs acts as an oncogenic signal promoting an exit from dormancy of therapy-induced cancer stem-like cells and leading to endocrine therapy resistance in OXPHOS-dependent breast cancer.
Keywords: cancer stem cells; exosomes; hormonal therapy; metastasis; mitochondrial DNA.
Conflict of interest statement
Conflict of interest statement: P.S. and J.B. hold a patent on this discovery (US 62/451,453).
Figures
References
-
- Fadoukhair Z, et al. Evaluation of targeted therapies in advanced breast cancer: The need for large-scale molecular screening and transformative clinical trial designs. Oncogene. 2016;35:1743–1749. - PubMed
-
- Ignatiadis M, Sotiriou C. Luminal breast cancer: From biology to treatment. Nat Rev Clin Oncol. 2013;10:494–506. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
