

Mitochondrial NAD+/NADH Redox State and Diabetic Cardiomyopathy
- PMID: 29073779
- PMCID: PMC6306679
- DOI: 10.1089/ars.2017.7415
Mitochondrial NAD+/NADH Redox State and Diabetic Cardiomyopathy
Abstract
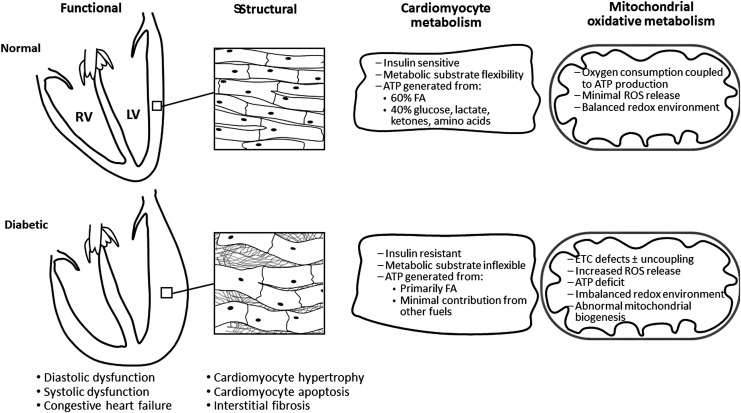
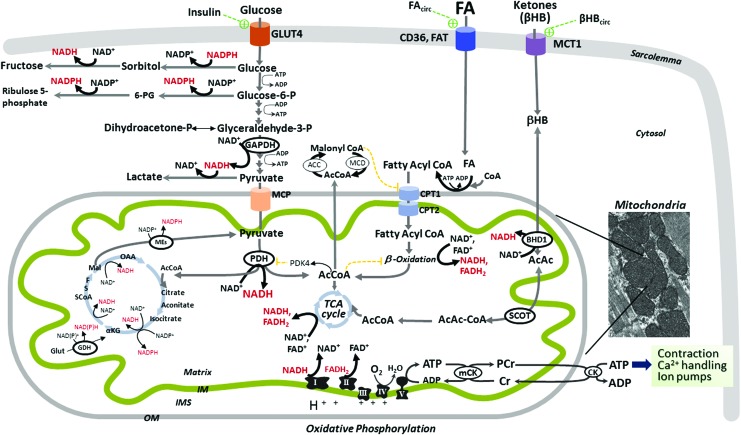
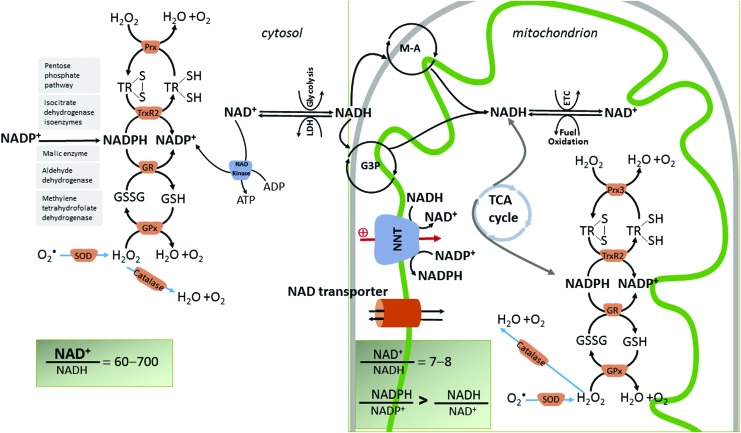
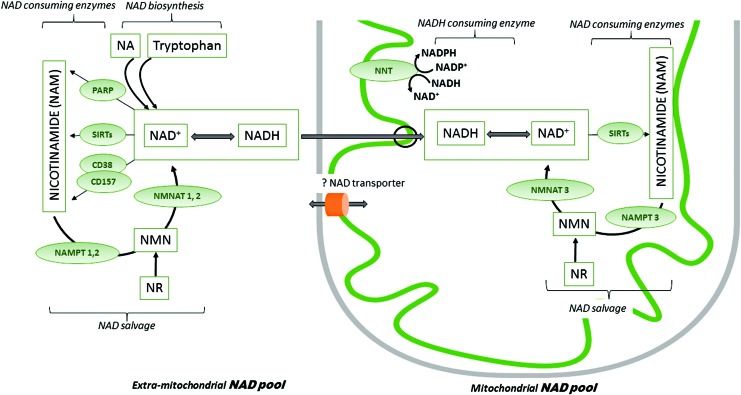
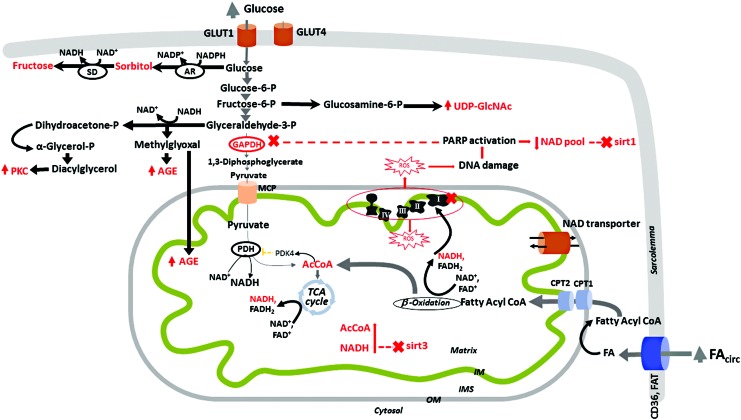
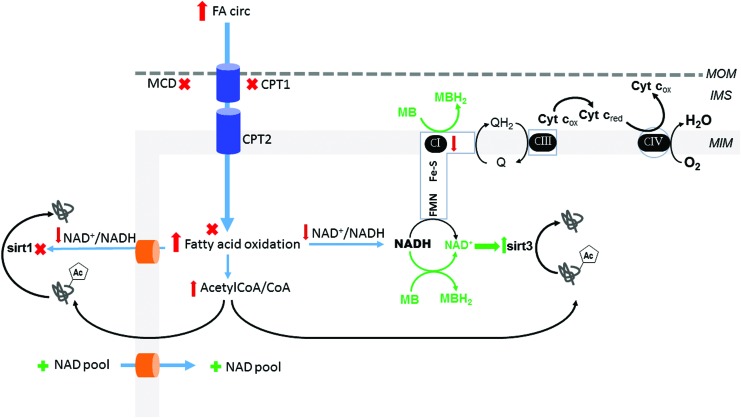
Significance: Diabetic cardiomyopathy (DCM) is a frequent complication occurring even in well-controlled asymptomatic diabetic patients, and it may advance to heart failure (HF). Recent Advances: The diabetic heart is characterized by a state of "metabolic rigidity" involving enhanced rates of fatty acid uptake and mitochondrial oxidation as the predominant energy source, and it exhibits mitochondrial electron transport chain defects. These alterations promote redox state changes evidenced by a decreased NAD+/NADH ratio associated with an increase in acetyl-CoA/CoA ratio. NAD+ is a co-substrate for deacetylases, sirtuins, and a critical molecule in metabolism and redox signaling; whereas acetyl-CoA promotes protein lysine acetylation, affecting mitochondrial integrity and causing epigenetic changes. Critical Issues: DCM lacks specific therapies with treatment only in later disease stages using standard, palliative HF interventions. Traditional therapy targeting neurohormonal signaling and hemodynamics failed to improve mortality rates. Though mitochondrial redox state changes occur in the heart with obesity and diabetes, how the mitochondrial NAD+/NADH redox couple connects the remodeled energy metabolism with mitochondrial and cytosolic antioxidant defense and nuclear epigenetic changes remains to be determined. Mitochondrial therapies targeting the mitochondrial NAD+/NADH redox ratio may alleviate cardiac dysfunction. Future Directions: Specific therapies must be supported by an optimal understanding of changes in mitochondrial redox state and how it influences other cellular compartments; this field has begun to surface as a therapeutic target for the diabetic heart. We propose an approach based on an alternate mitochondrial electron transport that normalizes the mitochondrial redox state and improves cardiac function in diabetes.
Keywords: NAD; NADH; cardiomyopathy; diabetes; mitochondria; redox balance.
Conflict of interest statement
The authors confirm that no competing financial interests exist. The authors apologize for not referring other studies due to the length constraints.
Figures
References
-
- Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, Minnemann T, Boers ME, Hadro E, Oberste-Berghaus C, Quist W, Lowell BB, Ingwall JS, and Kahn BB. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest 104: 1703–1714, 1999 - PMC - PubMed
-
- Alano CC, Tran A, Tao R, Ying W, Karliner JS, and Swanson RA. Differences among cell types in NAD(+) compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J Neurosci Res 85: 3378–3385, 2007 - PubMed
-
- Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, and Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res 100: 1512–1521, 2007 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous