

Autophagy and the unfolded protein response promote profibrotic effects of TGF-β1 in human lung fibroblasts
- PMID: 29074489
- PMCID: PMC5900356
- DOI: 10.1152/ajplung.00372.2017
Autophagy and the unfolded protein response promote profibrotic effects of TGF-β1 in human lung fibroblasts
Abstract
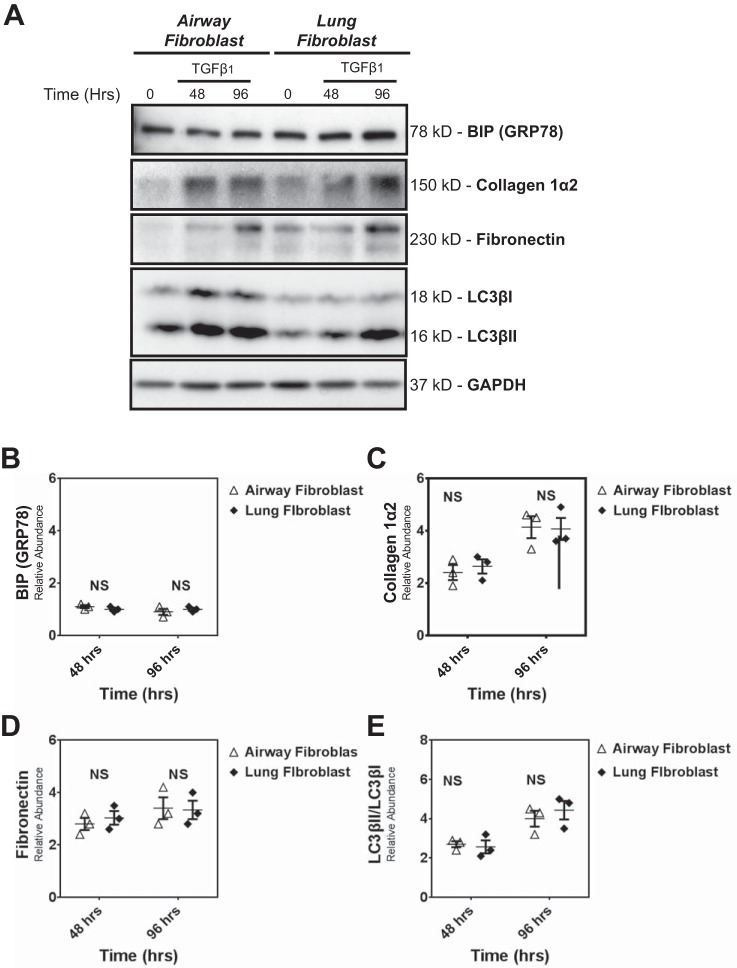
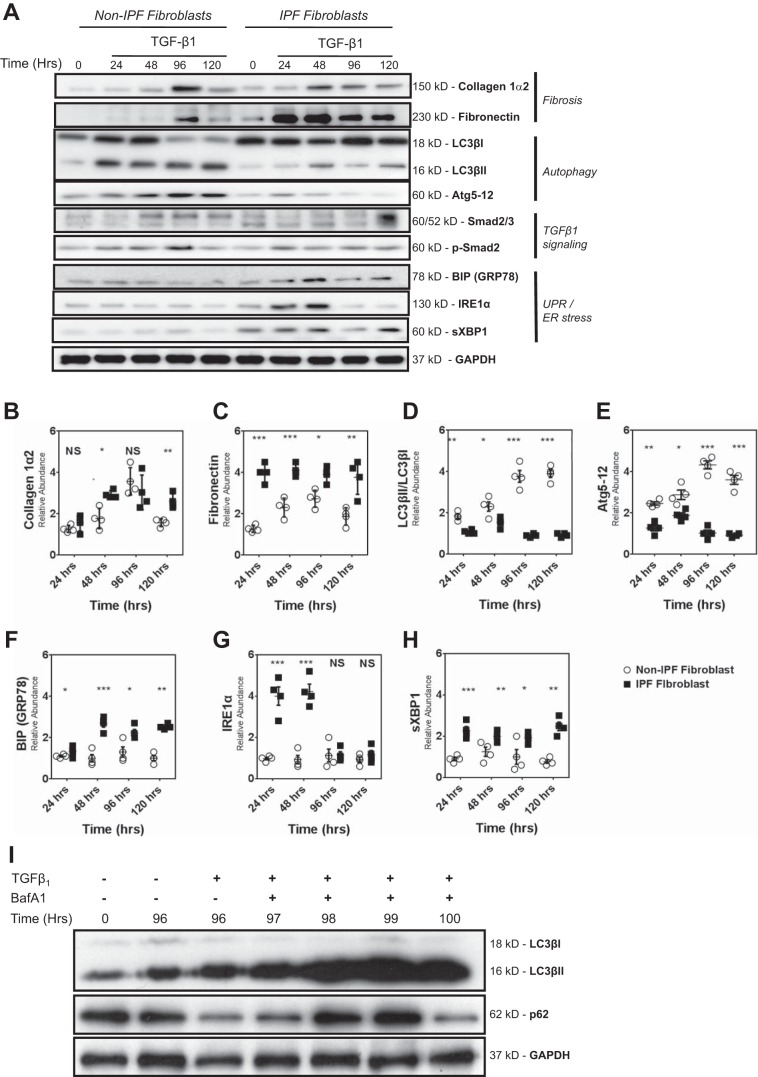
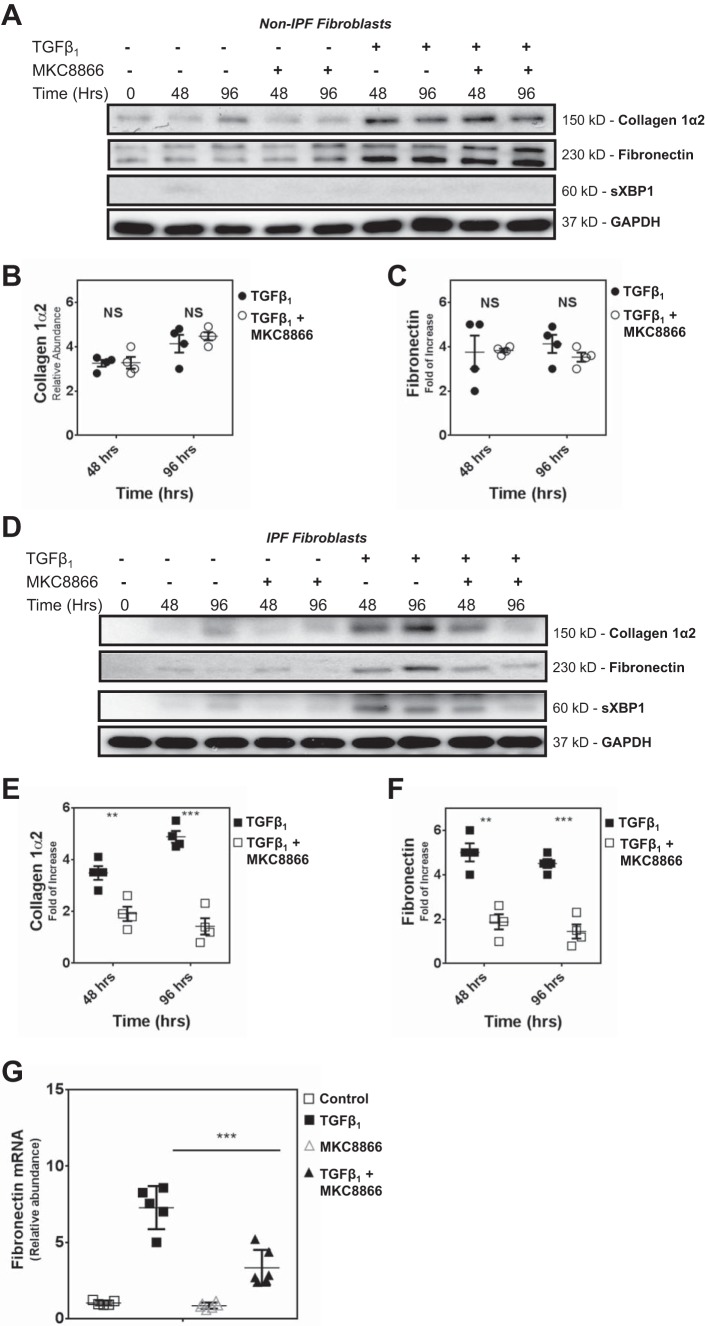
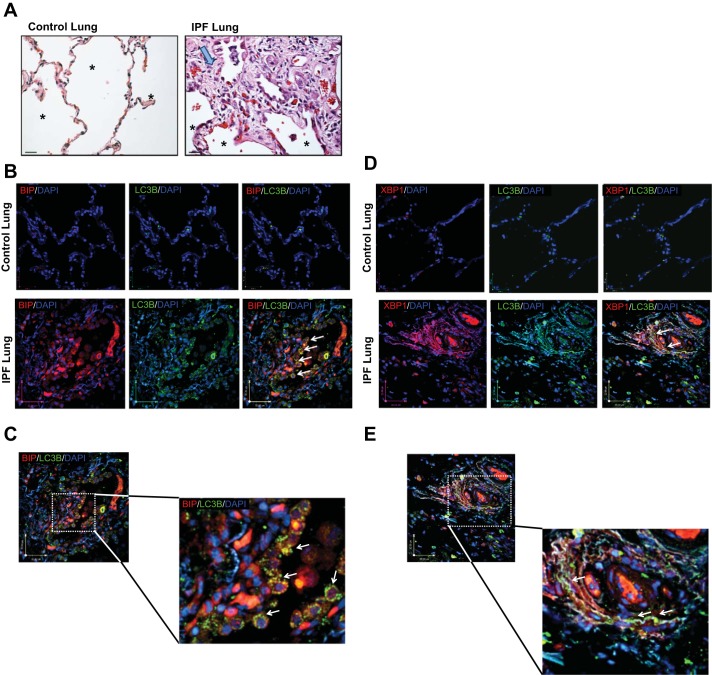
Idiopathic pulmonary fibrosis (IPF) is a lethal fibrotic lung disease in adults with limited treatment options. Autophagy and the unfolded protein response (UPR), fundamental processes induced by cell stress, are dysregulated in lung fibroblasts and epithelial cells from humans with IPF. Human primary cultured lung parenchymal and airway fibroblasts from non-IPF and IPF donors were stimulated with transforming growth factor-β1 (TGF-β1) with or without inhibitors of autophagy or UPR (IRE1 inhibitor). Using immunoblotting, we monitored temporal changes in abundance of protein markers of autophagy (LC3βII and Atg5-12), UPR (BIP, IRE1α, and cleaved XBP1), and fibrosis (collagen 1α2 and fibronectin). Using fluorescent immunohistochemistry, we profiled autophagy (LC3βII) and UPR (BIP and XBP1) markers in human non-IPF and IPF lung tissue. TGF-β1-induced collagen 1α2 and fibronectin protein production was significantly higher in IPF lung fibroblasts compared with lung and airway fibroblasts from non-IPF donors. TGF-β1 induced the accumulation of LC3βII in parallel with collagen 1α2 and fibronectin, but autophagy marker content was significantly lower in lung fibroblasts from IPF subjects. TGF-β1-induced collagen and fibronectin biosynthesis was significantly reduced by inhibiting autophagy flux in fibroblasts from the lungs of non-IPF and IPF donors. Conversely, only in lung fibroblasts from IPF donors did TGF-β1 induce UPR markers. Treatment with an IRE1 inhibitor decreased TGF-β1-induced collagen 1α2 and fibronectin biosynthesis in IPF lung fibroblasts but not those from non-IPF donors. The IRE1 arm of the UPR response is uniquely induced by TGF-β1 in lung fibroblasts from human IPF donors and is required for excessive biosynthesis of collagen and fibronectin in these cells.
Keywords: IRE1; pulmonary fibrosis; spliced XBP1; transforming growth factor-β1.
Figures


References
-
- Adamson YI, Bowden DH. Pulmonary injury and repair. Organ culture studies of murine lung after oxygen. Arch Pathol Lab Med 100: 640–643, 1976. - PubMed
-
- Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Kawabata Y, Hano H, Nakayama K, Kuwano K. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 304: L56–L69, 2013. doi: 10.1152/ajplung.00213.2012. - DOI - PubMed
-
- Borges FT, Melo SA, Özdemir BC, Kato N, Revuelta I, Miller CA, Gattone VH II, LeBleu VS, Kalluri R. TGF-β1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J Am Soc Nephrol 24: 385–392, 2013. doi: 10.1681/ASN.2012101031. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous