

Efficient Prioritization of Multiple Causal eQTL Variants via Sparse Polygenic Modeling
- PMID: 29074555
- PMCID: PMC5714449
- DOI: 10.1534/genetics.117.300435
Efficient Prioritization of Multiple Causal eQTL Variants via Sparse Polygenic Modeling
Abstract
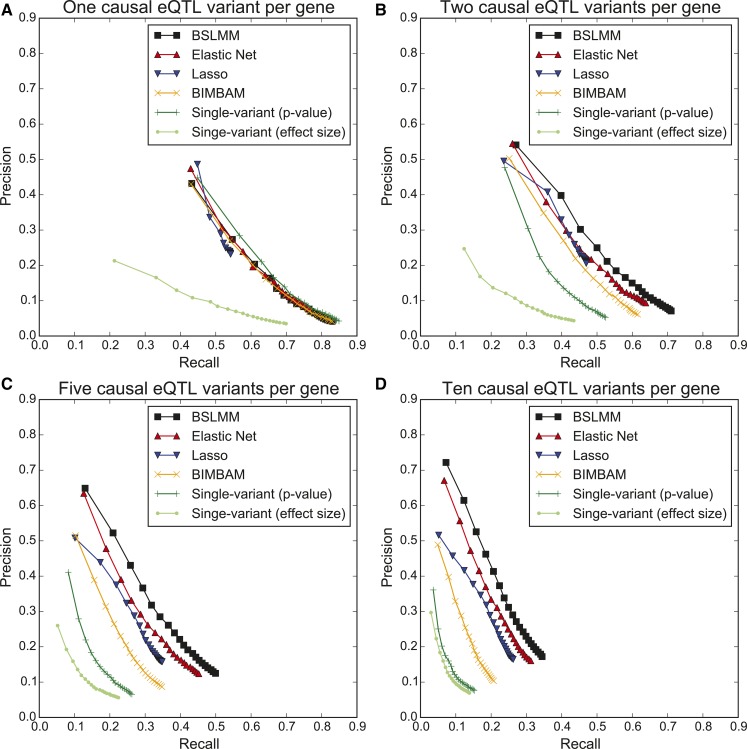
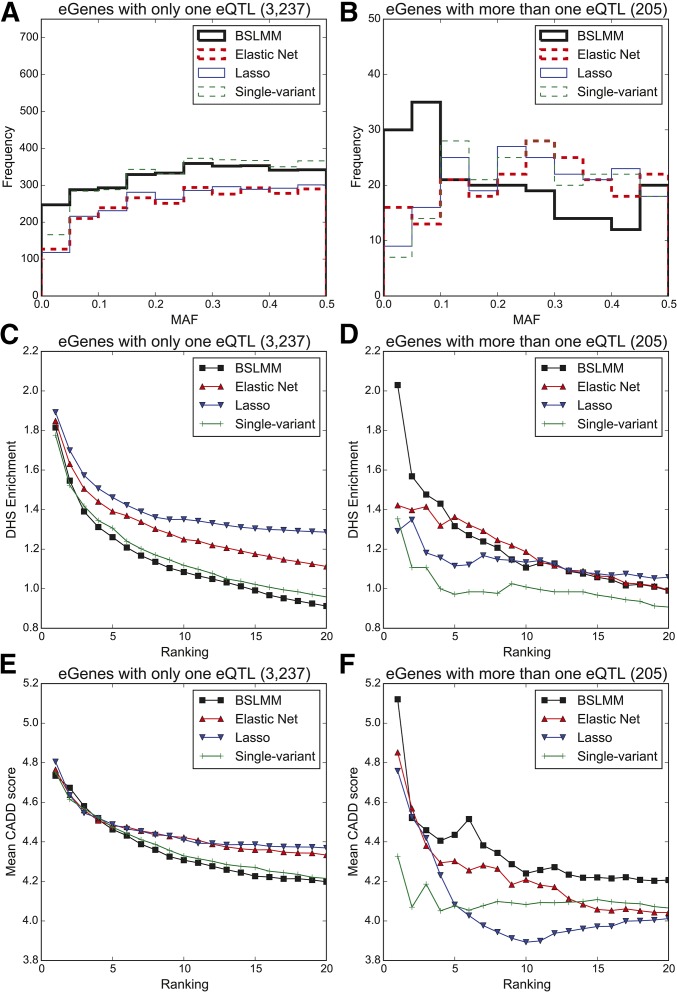
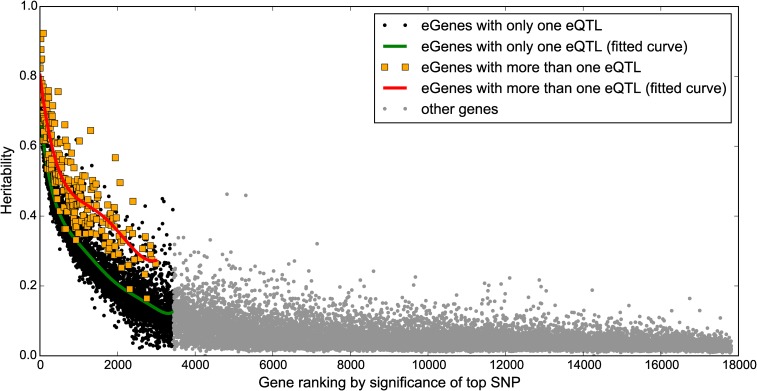
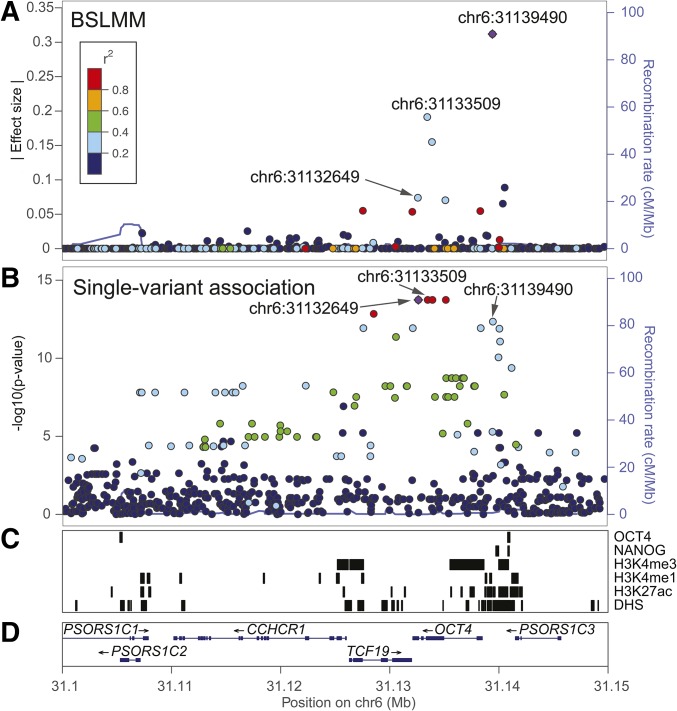
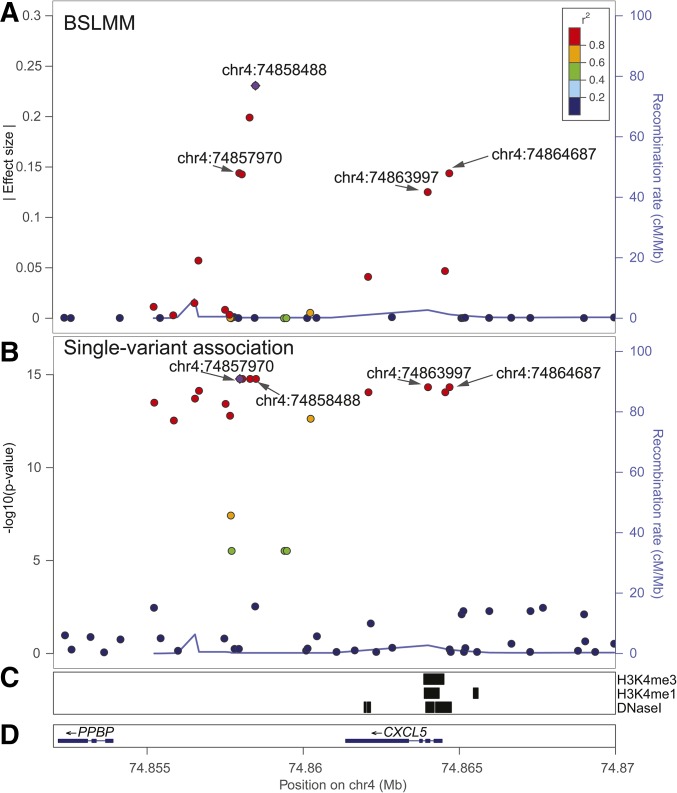
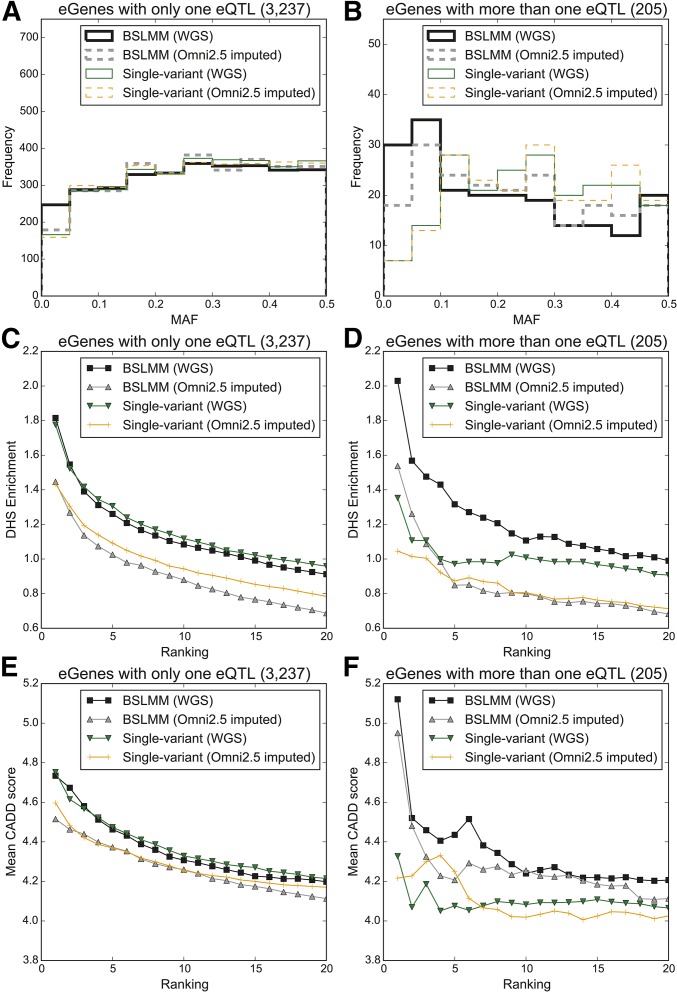
Expression quantitative trait loci (eQTL) studies have typically used single-variant association analysis to identify genetic variants correlated with gene expression. However, this approach has several drawbacks: causal variants cannot be distinguished from nonfunctional variants in strong linkage disequilibrium, combined effects from multiple causal variants cannot be captured, and low-frequency (<5% MAF) eQTL variants are difficult to identify. While these issues possibly could be overcome by using sparse polygenic models, which associate multiple genetic variants with gene expression simultaneously, the predictive performance of these models for eQTL studies has not been evaluated. Here, we assessed the ability of three sparse polygenic models (Lasso, Elastic Net, and BSLMM) to identify causal variants, and compared their efficacy to single-variant association analysis and a fine-mapping model. Using simulated data, we determined that, while these methods performed similarly when there was one causal SNP present at a gene, BSLMM substantially outperformed single-variant association analysis for prioritizing causal eQTL variants when multiple causal eQTL variants were present (1.6- to 5.2-fold higher recall at 20% precision), and identified up to 2.3-fold more low frequency variants as the top eQTL SNP. Analysis of real RNA-seq and whole-genome sequencing data of 131 iPSC samples showed that the eQTL SNPs identified by BSLMM had a higher functional enrichment in DHS sites and were more often low-frequency than those identified with single-variant association analysis. Our study showed that BSLMM is a more effective approach than single-variant association analysis for prioritizing multiple causal eQTL variants at a single gene.
Keywords: causal variants; eQTLs; sparse polygenic models.
Copyright © 2017 by the Genetics Society of America.
Figures
Similar articles
-
Leveraging allelic imbalance to refine fine-mapping for eQTL studies.PLoS Genet. 2019 Dec 13;15(12):e1008481. doi: 10.1371/journal.pgen.1008481. eCollection 2019 Dec. PLoS Genet. 2019. PMID: 31834882 Free PMC article.
-
Genetic control of gene expression at novel and established chronic obstructive pulmonary disease loci.Hum Mol Genet. 2015 Feb 15;24(4):1200-10. doi: 10.1093/hmg/ddu525. Epub 2014 Oct 14. Hum Mol Genet. 2015. PMID: 25315895 Free PMC article.
-
Endometrial vezatin and its association with endometriosis risk.Hum Reprod. 2016 May;31(5):999-1013. doi: 10.1093/humrep/dew047. Epub 2016 Mar 22. Hum Reprod. 2016. PMID: 27005890
-
Expression Quantitative Trait Loci Information Improves Predictive Modeling of Disease Relevance of Non-Coding Genetic Variation.PLoS One. 2015 Oct 16;10(10):e0140758. doi: 10.1371/journal.pone.0140758. eCollection 2015. PLoS One. 2015. PMID: 26474488 Free PMC article. Review.
-
Expression QTLs Mapping and Analysis: A Bayesian Perspective.Methods Mol Biol. 2017;1488:189-215. doi: 10.1007/978-1-4939-6427-7_8. Methods Mol Biol. 2017. PMID: 27933525 Review.
Cited by
-
Sex-heterogeneous SNPs disproportionately influence gene expression and health.PLoS Genet. 2022 May 5;18(5):e1010147. doi: 10.1371/journal.pgen.1010147. eCollection 2022 May. PLoS Genet. 2022. PMID: 35511767 Free PMC article.
-
SNP eQTL status and eQTL density in the adjacent region of the SNP are associated with its statistical significance in GWA studies.BMC Genet. 2019 Nov 12;20(1):85. doi: 10.1186/s12863-019-0786-0. BMC Genet. 2019. PMID: 31718536 Free PMC article.
-
Biological relevance of computationally predicted pathogenicity of noncoding variants.Nat Commun. 2019 Jan 18;10(1):330. doi: 10.1038/s41467-018-08270-y. Nat Commun. 2019. PMID: 30659175 Free PMC article.
-
Combining artificial intelligence: deep learning with Hi-C data to predict the functional effects of non-coding variants.Bioinformatics. 2021 Jun 16;37(10):1339-1344. doi: 10.1093/bioinformatics/btaa970. Bioinformatics. 2021. PMID: 33196774 Free PMC article.
-
Fine mapping spatiotemporal mechanisms of genetic variants underlying cardiac traits and disease.Nat Commun. 2023 Feb 28;14(1):1132. doi: 10.1038/s41467-023-36638-2. Nat Commun. 2023. PMID: 36854752 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
