

Deep sequencing of natural and experimental populations of Drosophila melanogaster reveals biases in the spectrum of new mutations
- PMID: 29079675
- PMCID: PMC5741049
- DOI: 10.1101/gr.219956.116
Deep sequencing of natural and experimental populations of Drosophila melanogaster reveals biases in the spectrum of new mutations
Abstract
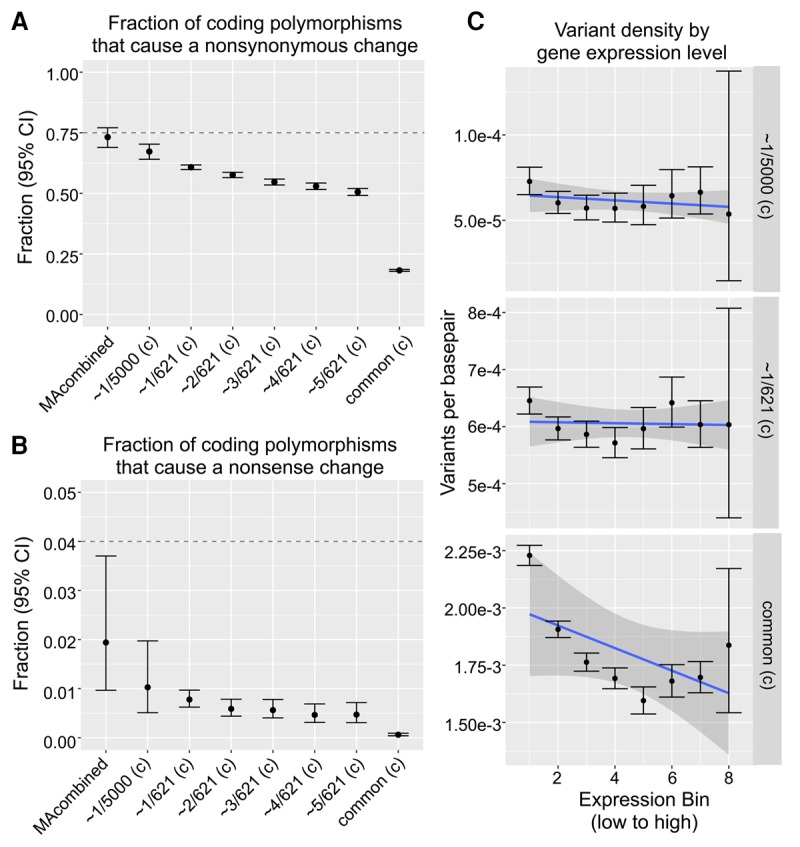
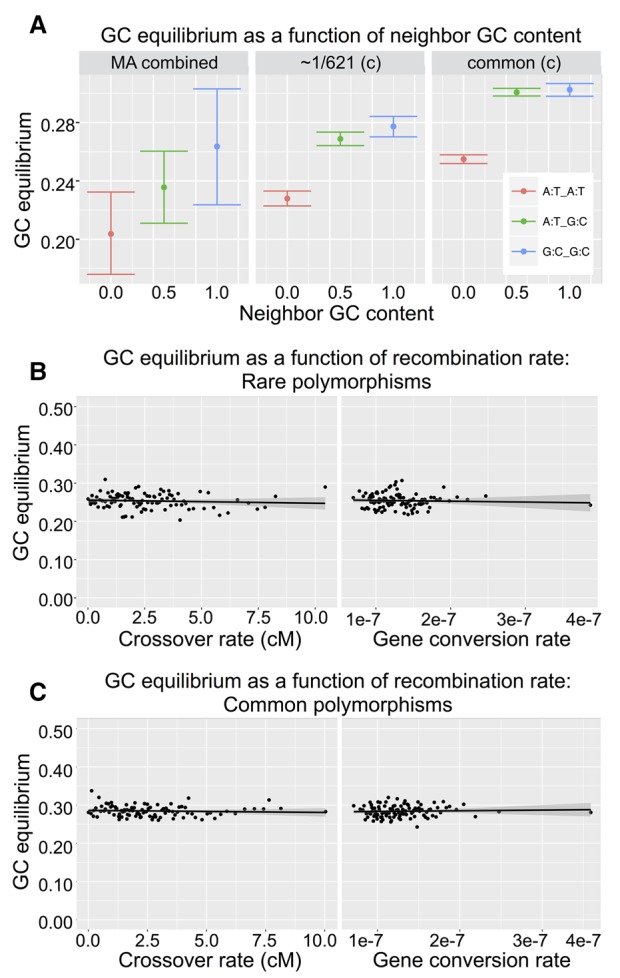
Mutations provide the raw material of evolution, and thus our ability to study evolution depends fundamentally on having precise measurements of mutational rates and patterns. We generate a data set for this purpose using (1) de novo mutations from mutation accumulation experiments and (2) extremely rare polymorphisms from natural populations. The first, mutation accumulation (MA) lines are the product of maintaining flies in tiny populations for many generations, therefore rendering natural selection ineffective and allowing new mutations to accrue in the genome. The second, rare genetic variation from natural populations allows the study of mutation because extremely rare polymorphisms are relatively unaffected by the filter of natural selection. We use both methods in Drosophila melanogaster, first generating our own novel data set of sequenced MA lines and performing a meta-analysis of all published MA mutations (∼2000 events) and then identifying a high quality set of ∼70,000 extremely rare (≤0.1%) polymorphisms that are fully validated with resequencing. We use these data sets to precisely measure mutational rates and patterns. Highlights of our results include: a high rate of multinucleotide mutation events at both short (∼5 bp) and long (∼1 kb) genomic distances, showing that mutation drives GC content lower in already GC-poor regions, and using our precise context-dependent mutation rates to predict long-term evolutionary patterns at synonymous sites. We also show that de novo mutations from independent MA experiments display similar patterns of single nucleotide mutation and well match the patterns of mutation found in natural populations.
© 2017 Assaf et al.; Published by Cold Spring Harbor Laboratory Press.
Figures





References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous