

ARSDA: A New Approach for Storing, Transmitting and Analyzing Transcriptomic Data
- PMID: 29079682
- PMCID: PMC5714481
- DOI: 10.1534/g3.117.300271
ARSDA: A New Approach for Storing, Transmitting and Analyzing Transcriptomic Data
Abstract
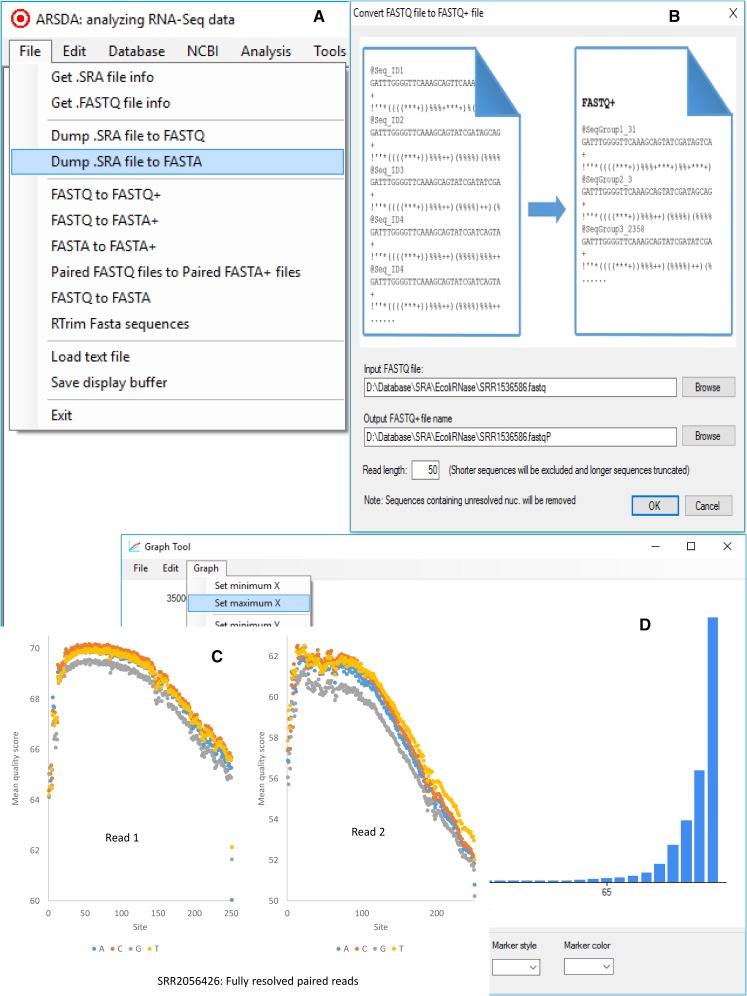
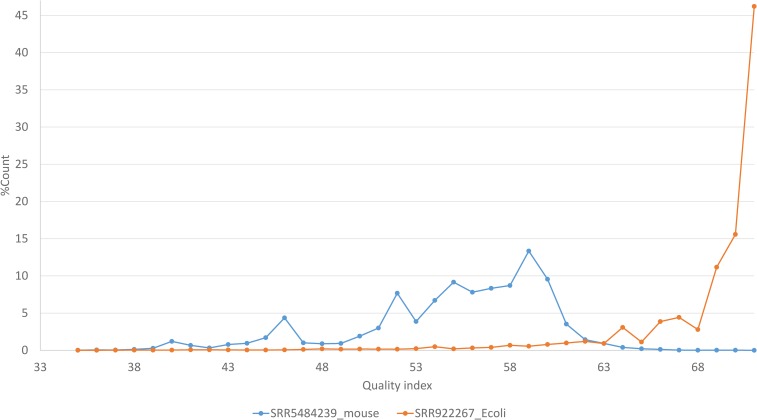
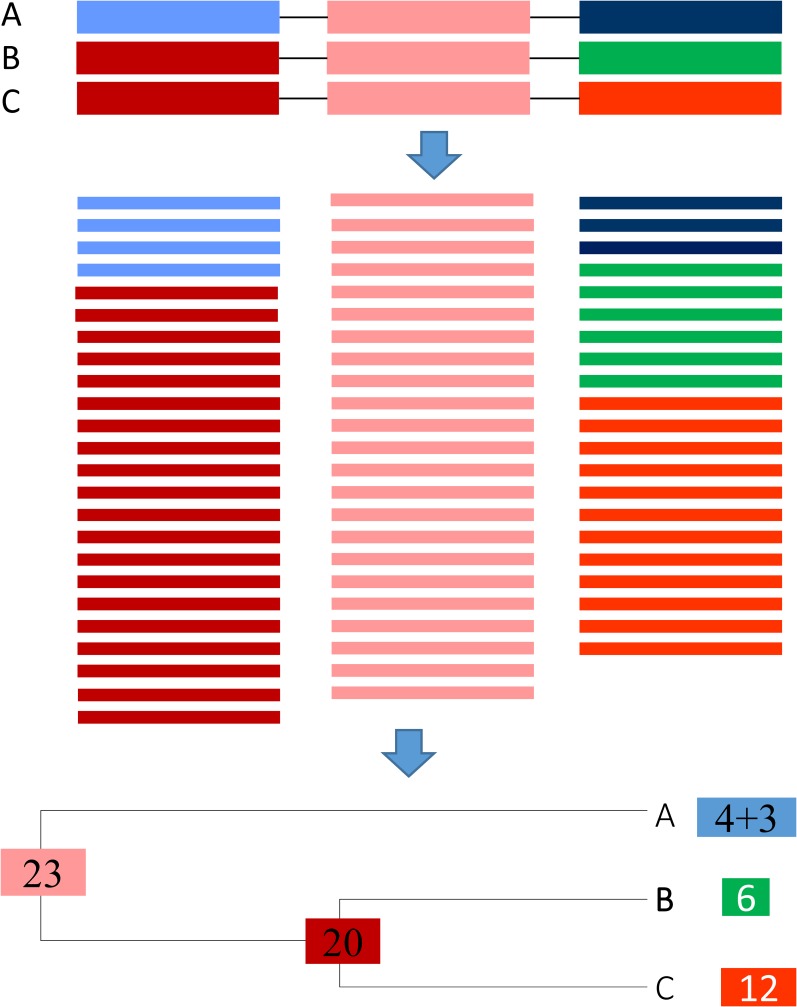
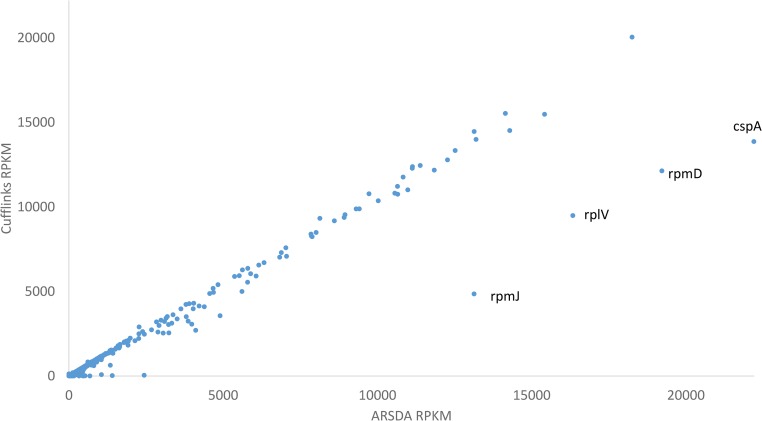
Two major stumbling blocks exist in high-throughput sequencing (HTS) data analysis. The first is the sheer file size, typically in gigabytes when uncompressed, causing problems in storage, transmission, and analysis. However, these files do not need to be so large, and can be reduced without loss of information. Each HTS file, either in compressed .SRA or plain text .fastq format, contains numerous identical reads stored as separate entries. For example, among 44,603,541 forward reads in the SRR4011234.sra file (from a Bacillus subtilis transcriptomic study) deposited at NCBI's SRA database, one read has 497,027 identical copies. Instead of storing them as separate entries, one can and should store them as a single entry with the SeqID_NumCopy format (which I dub as FASTA+ format). The second is the proper allocation of reads that map equally well to paralogous genes. I illustrate in detail a new method for such allocation. I have developed ARSDA software that implement these new approaches. A number of HTS files for model species are in the process of being processed and deposited at http://coevol.rdc.uottawa.ca to demonstrate that this approach not only saves a huge amount of storage space and transmission bandwidth, but also dramatically reduces time in downstream data analysis. Instead of matching the 497,027 identical reads separately against the B. subtilis genome, one only needs to match it once. ARSDA includes functions to take advantage of HTS data in the new sequence format for downstream data analysis such as gene expression characterization. I contrasted gene expression results between ARSDA and Cufflinks so readers can better appreciate the strength of ARSDA. ARSDA is freely available for Windows, Linux. and Macintosh computers at http://dambe.bio.uottawa.ca/ARSDA/ARSDA.aspx.
Keywords: ARSDA; novel storage solution; quantifying expression of paralogous genes; sequence format; transcriptomics.
Copyright © 2017 Xia.
Figures

Similar articles
-
Broom: application for non-redundant storage of high throughput sequencing data.Bioinformatics. 2019 Jan 1;35(1):143-145. doi: 10.1093/bioinformatics/bty580. Bioinformatics. 2019. PMID: 30010786
-
BEETL-fastq: a searchable compressed archive for DNA reads.Bioinformatics. 2014 Oct;30(19):2796-801. doi: 10.1093/bioinformatics/btu387. Epub 2014 Jun 20. Bioinformatics. 2014. PMID: 24950811
-
Reference-based compression of short-read sequences using path encoding.Bioinformatics. 2015 Jun 15;31(12):1920-8. doi: 10.1093/bioinformatics/btv071. Epub 2015 Feb 2. Bioinformatics. 2015. PMID: 25649622 Free PMC article.
-
Single-cell and spatial transcriptomics: Bridging current technologies with long-read sequencing.Mol Aspects Med. 2024 Apr;96:101255. doi: 10.1016/j.mam.2024.101255. Epub 2024 Feb 17. Mol Aspects Med. 2024. PMID: 38368637 Review.
-
The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants.Nucleic Acids Res. 2010 Apr;38(6):1767-71. doi: 10.1093/nar/gkp1137. Epub 2009 Dec 16. Nucleic Acids Res. 2010. PMID: 20015970 Free PMC article. Review.
Cited by
-
A computational system for identifying operons based on RNA-seq data.Methods. 2020 Apr 1;176:62-70. doi: 10.1016/j.ymeth.2019.03.026. Epub 2019 Apr 4. Methods. 2020. PMID: 30953757 Free PMC article. Review.
-
An improved estimation of tRNA expression to better elucidate the coevolution between tRNA abundance and codon usage in bacteria.Sci Rep. 2019 Feb 28;9(1):3184. doi: 10.1038/s41598-019-39369-x. Sci Rep. 2019. PMID: 30816249 Free PMC article.
-
Does Saccharomyces cerevisiae Require Specific Post-Translational Silencing against Leaky Translation of Hac1up?Microorganisms. 2021 Mar 17;9(3):620. doi: 10.3390/microorganisms9030620. Microorganisms. 2021. PMID: 33802931 Free PMC article.
-
How Changes in Anti-SD Sequences Would Affect SD Sequences in Escherichia coli and Bacillus subtilis.G3 (Bethesda). 2017 May 5;7(5):1607-1615. doi: 10.1534/g3.117.039305. G3 (Bethesda). 2017. PMID: 28364038 Free PMC article.
-
RNA-Seq-Based Analysis Reveals Heterogeneity in Mature 16S rRNA 3' Termini and Extended Anti-Shine-Dalgarno Motifs in Bacterial Species.G3 (Bethesda). 2018 Dec 10;8(12):3973-3979. doi: 10.1534/g3.118.200729. G3 (Bethesda). 2018. PMID: 30355764 Free PMC article.
References
-
- Abraham J. M., Feagin J. E., Stuart K., 1988. Characterization of cytochrome c oxidase III transcripts that are edited only in the 3′ region. Cell 55: 267–272. - PubMed
-
- Andrews, S., 2017 FastQC, Babraham Bioinformatics. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources