

Pharmacological inhibition of the transcription factor PU.1 in leukemia
- PMID: 29083320
- PMCID: PMC5707147
- DOI: 10.1172/JCI92504
Pharmacological inhibition of the transcription factor PU.1 in leukemia
Abstract
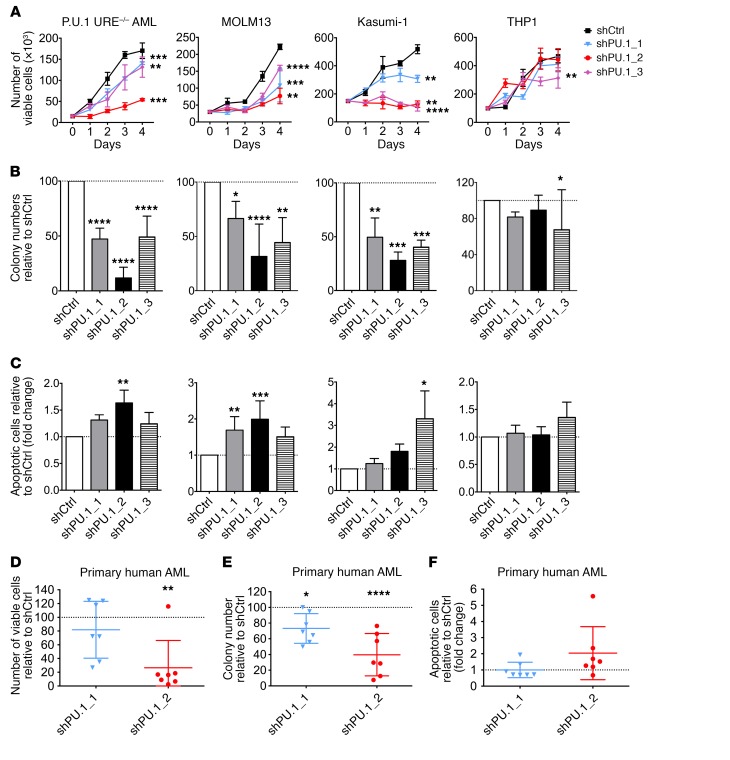
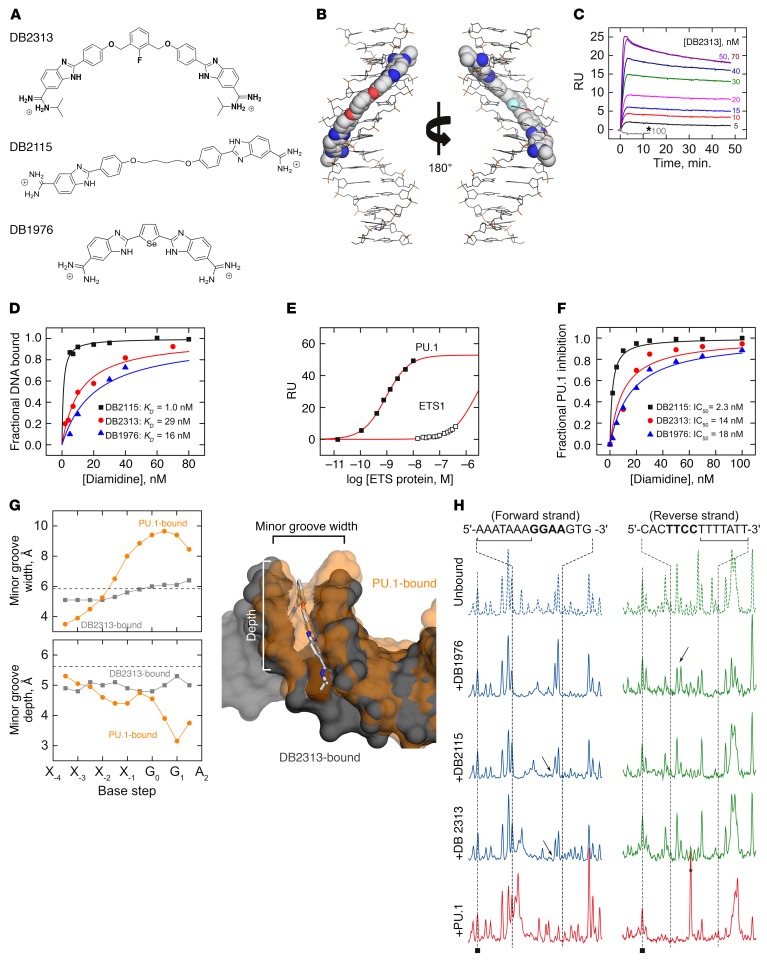
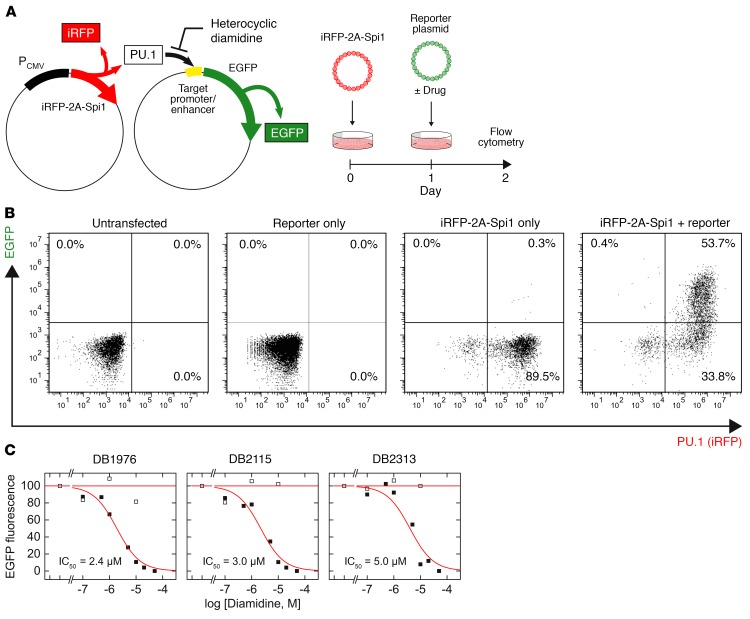
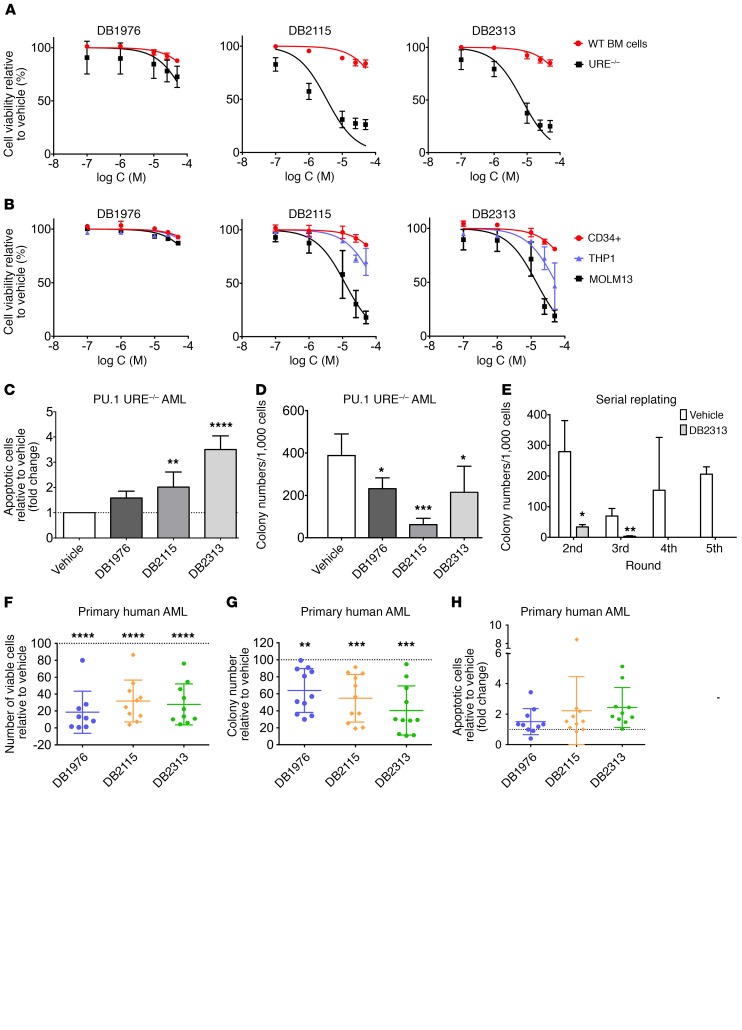
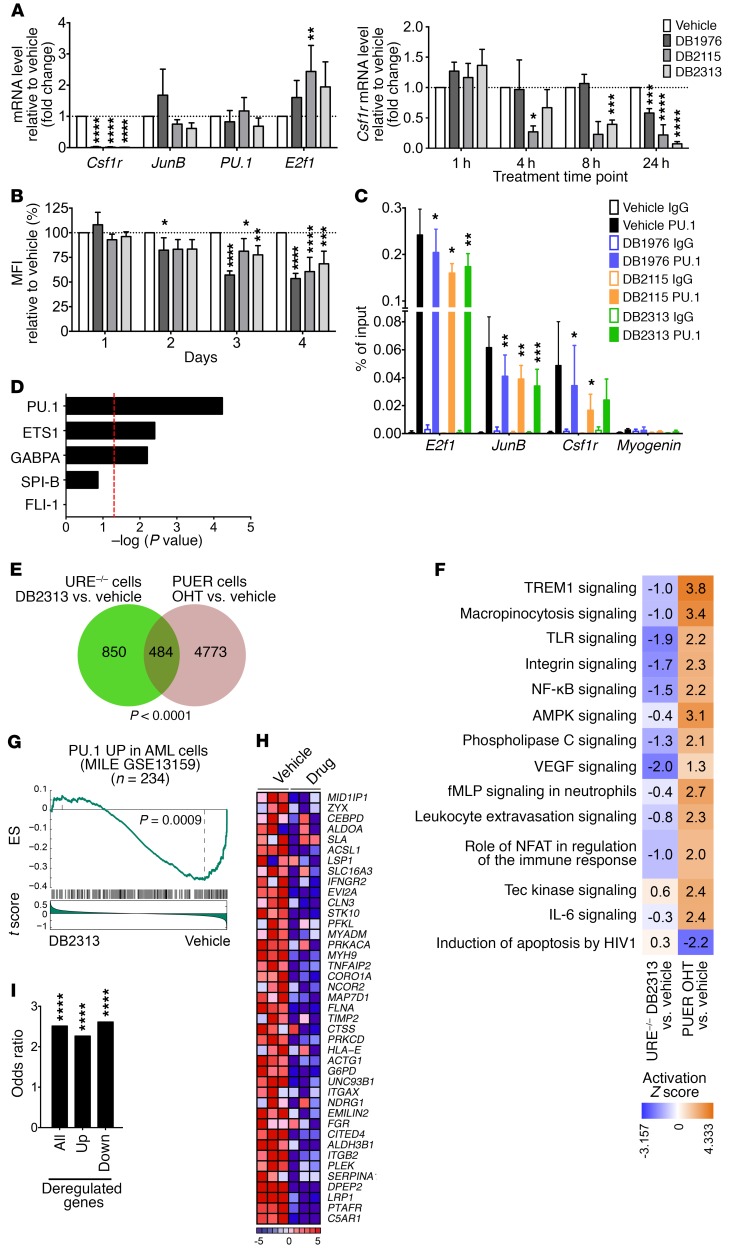
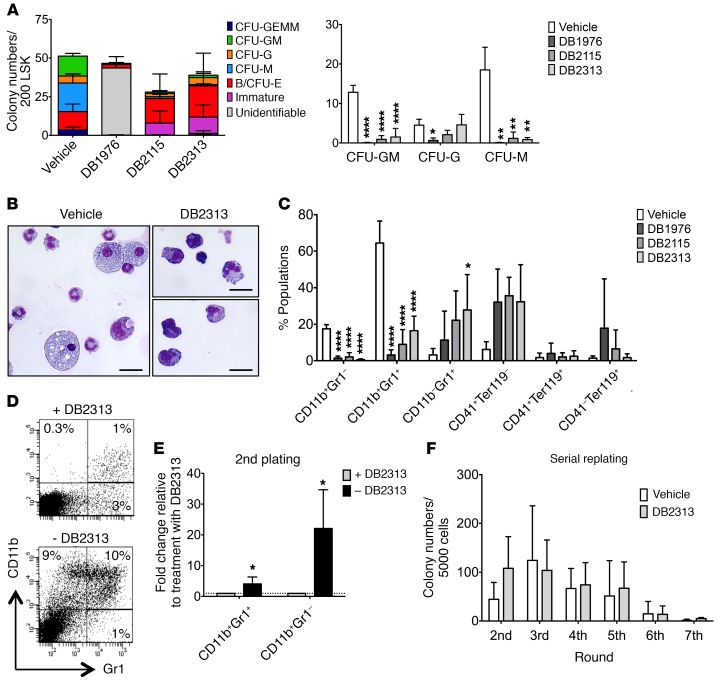
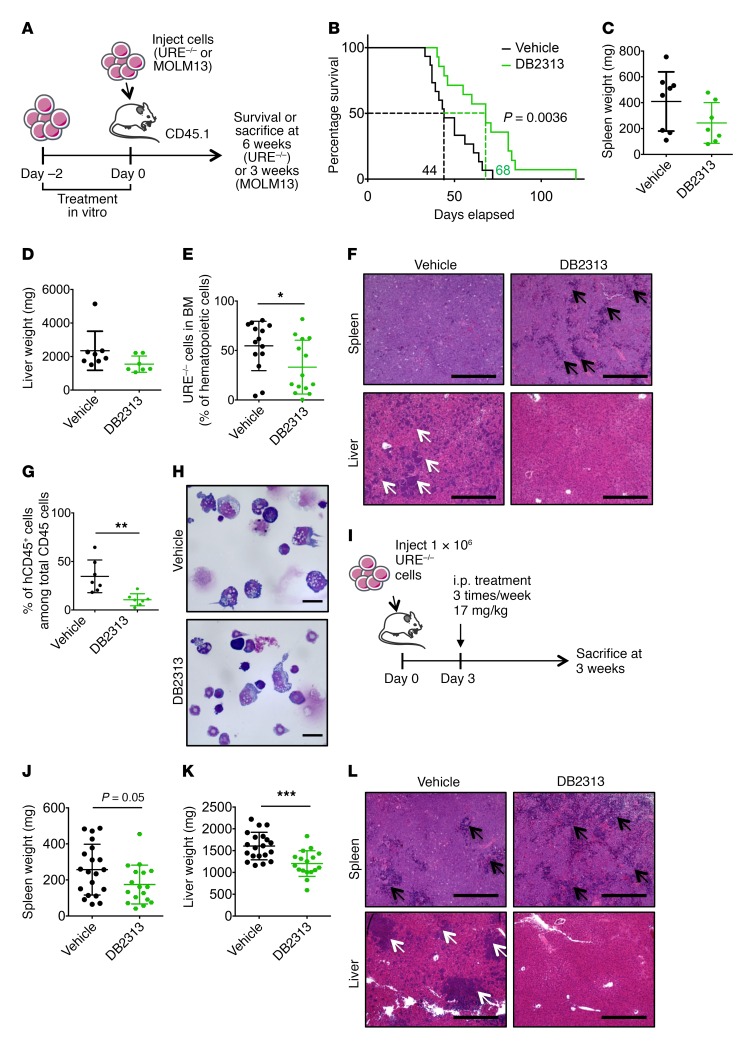
The transcription factor PU.1 is often impaired in patients with acute myeloid leukemia (AML). Here, we used AML cells that already had low PU.1 levels and further inhibited PU.1 using either RNA interference or, to our knowledge, first-in-class small-molecule inhibitors of PU.1 that we developed specifically to allosterically interfere with PU.1-chromatin binding through interaction with the DNA minor groove that flanks PU.1-binding motifs. These small molecules of the heterocyclic diamidine family disrupted the interaction of PU.1 with target gene promoters and led to downregulation of canonical PU.1 transcriptional targets. shRNA or small-molecule inhibition of PU.1 in AML cells from either PU.1lo mutant mice or human patients with AML-inhibited cell growth and clonogenicity and induced apoptosis. In murine and human AML (xeno)transplantation models, treatment with our PU.1 inhibitors decreased tumor burden and resulted in increased survival. Thus, our study provides proof of concept that PU.1 inhibition has potential as a therapeutic strategy for the treatment of AML and for the development of small-molecule inhibitors of PU.1.
Keywords: Drug therapy; Hematology; Leukemias; Therapeutics; Transcription.
Conflict of interest statement
Figures
References
-
- Facts 2014-2015. Leukemia & Lymphoma Society. https://www.lls.org/sites/default/files/file_assets/facts.pdf Accessed September 28, 2017.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
