

Maximum entropy methods for extracting the learned features of deep neural networks
- PMID: 29084280
- PMCID: PMC5679649
- DOI: 10.1371/journal.pcbi.1005836
Maximum entropy methods for extracting the learned features of deep neural networks
Abstract
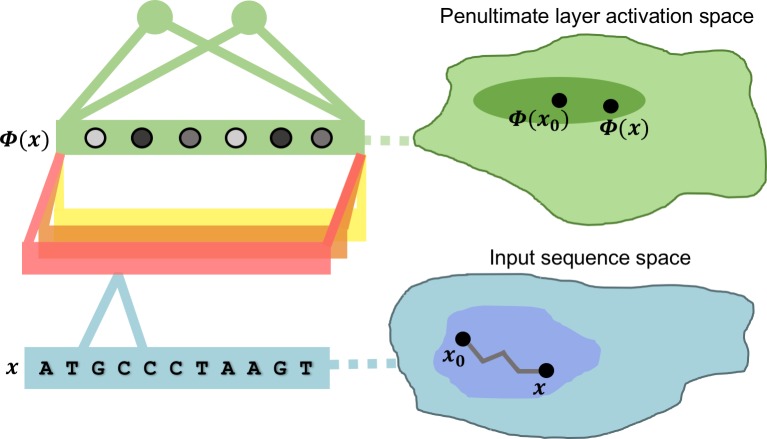
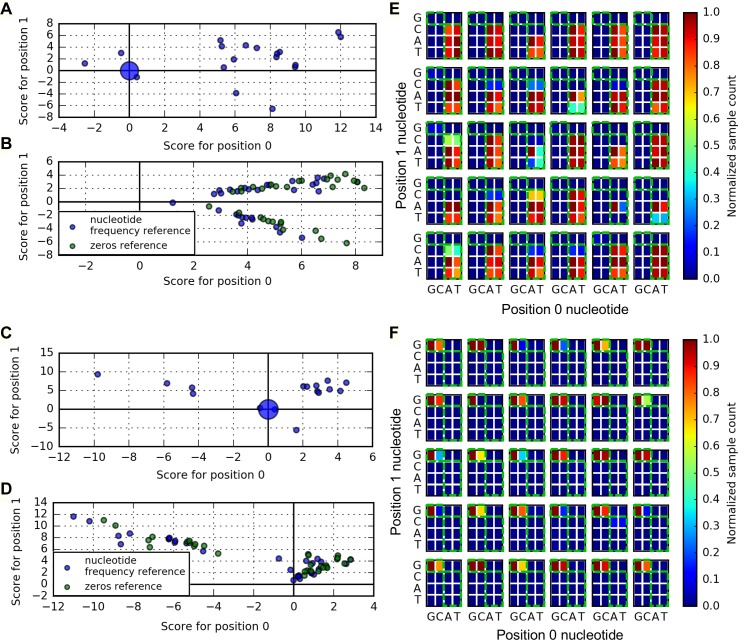
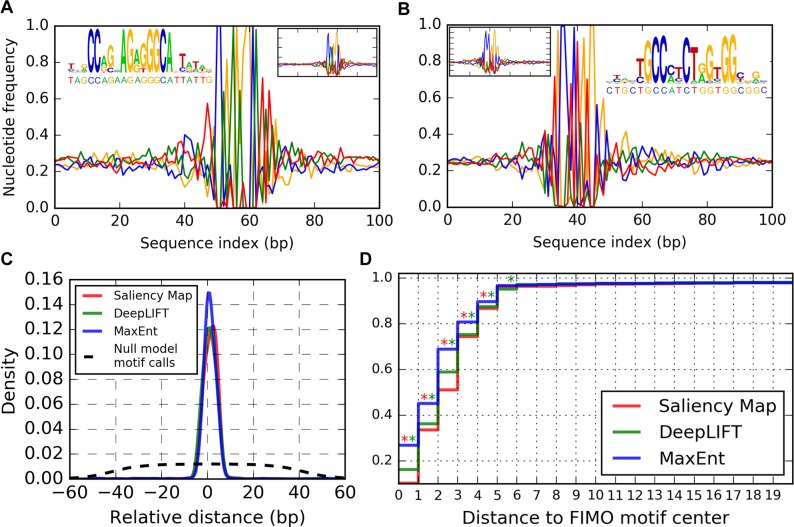
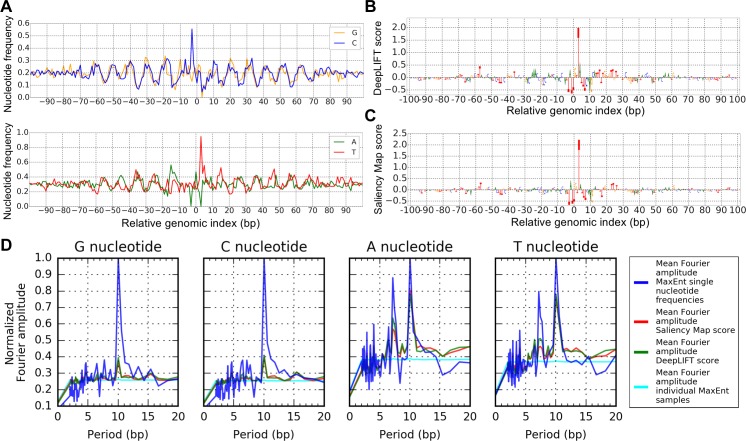
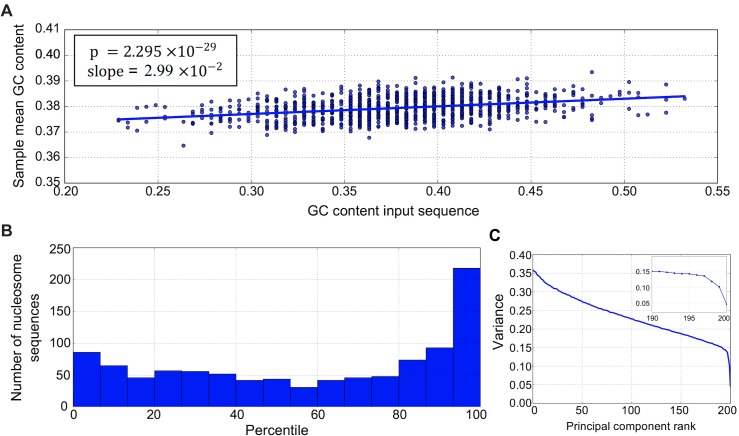
New architectures of multilayer artificial neural networks and new methods for training them are rapidly revolutionizing the application of machine learning in diverse fields, including business, social science, physical sciences, and biology. Interpreting deep neural networks, however, currently remains elusive, and a critical challenge lies in understanding which meaningful features a network is actually learning. We present a general method for interpreting deep neural networks and extracting network-learned features from input data. We describe our algorithm in the context of biological sequence analysis. Our approach, based on ideas from statistical physics, samples from the maximum entropy distribution over possible sequences, anchored at an input sequence and subject to constraints implied by the empirical function learned by a network. Using our framework, we demonstrate that local transcription factor binding motifs can be identified from a network trained on ChIP-seq data and that nucleosome positioning signals are indeed learned by a network trained on chemical cleavage nucleosome maps. Imposing a further constraint on the maximum entropy distribution also allows us to probe whether a network is learning global sequence features, such as the high GC content in nucleosome-rich regions. This work thus provides valuable mathematical tools for interpreting and extracting learned features from feed-forward neural networks.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Alipanahi B, Delong A, Weirauch MT, Frey BJ. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol. 2015;33(8):831–8. doi: 10.1038/nbt.3300 . - DOI - PubMed
-
- Zeng H, Edwards MD, Liu G, Gifford DK. Convolutional neural network architectures for predicting DNA-protein binding. Bioinformatics. 2016;32(12):i121–i7. doi: 10.1093/bioinformatics/btw255 ; PubMed Central PMCID: PMCPMC4908339. - DOI - PMC - PubMed
-
- Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods. 2015;12(10):931–4. doi: 10.1038/nmeth.3547 ; PubMed Central PMCID: PMCPMC4768299. - DOI - PMC - PubMed
-
- Kelley DR, Snoek J, Rinn JL. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016;26(7):990–9. doi: 10.1101/gr.200535.115 ; PubMed Central PMCID: PMCPMC4937568. - DOI - PMC - PubMed
-
- Zeng H, Gifford DK. Predicting the impact of non-coding variants on DNA methylation. Nucleic acids research. 2017. doi: 10.1093/nar/gkx177 . - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
