

The fitness cost of mis-splicing is the main determinant of alternative splicing patterns
- PMID: 29084568
- PMCID: PMC5663052
- DOI: 10.1186/s13059-017-1344-6
The fitness cost of mis-splicing is the main determinant of alternative splicing patterns
Abstract
Background: Most eukaryotic genes are subject to alternative splicing (AS), which may contribute to the production of protein variants or to the regulation of gene expression via nonsense-mediated messenger RNA (mRNA) decay (NMD). However, a fraction of splice variants might correspond to spurious transcripts and the question of the relative proportion of splicing errors to functional splice variants remains highly debated.
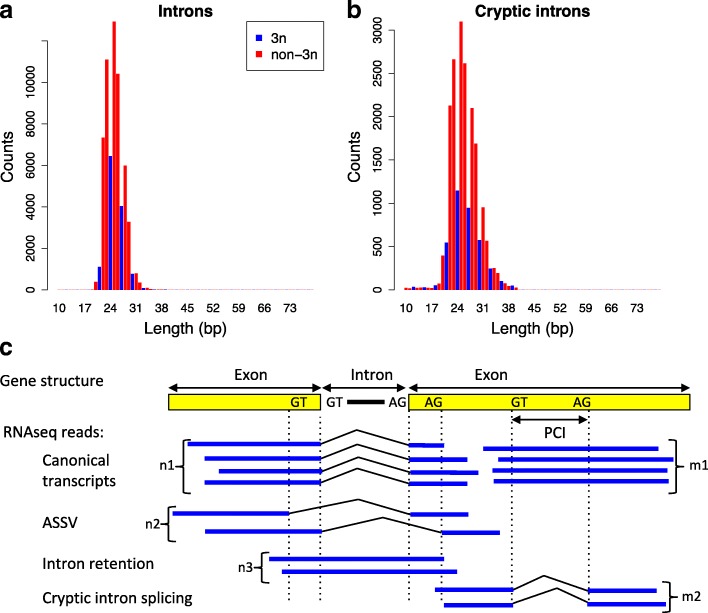
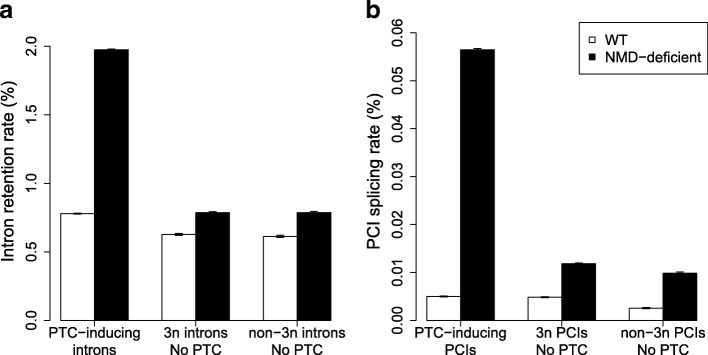
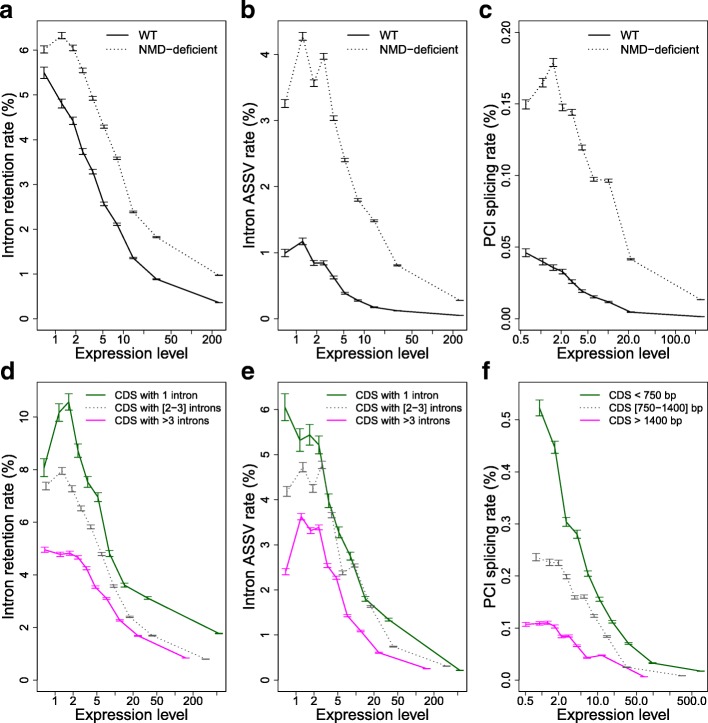
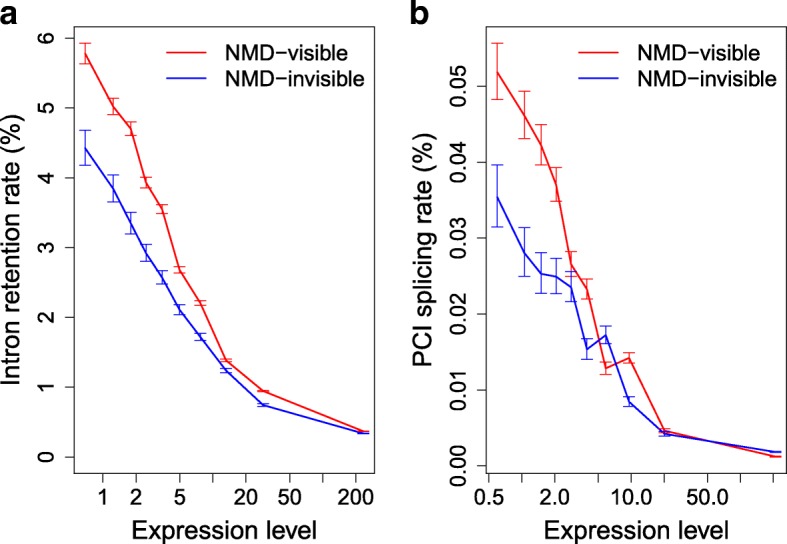
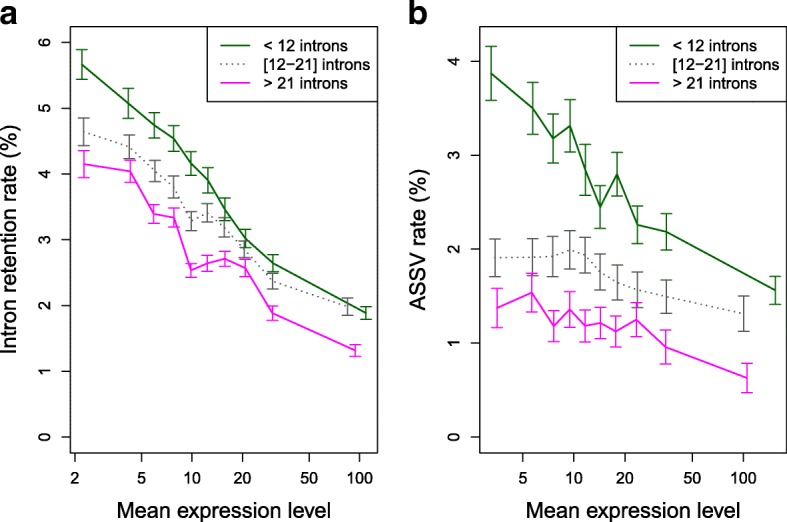
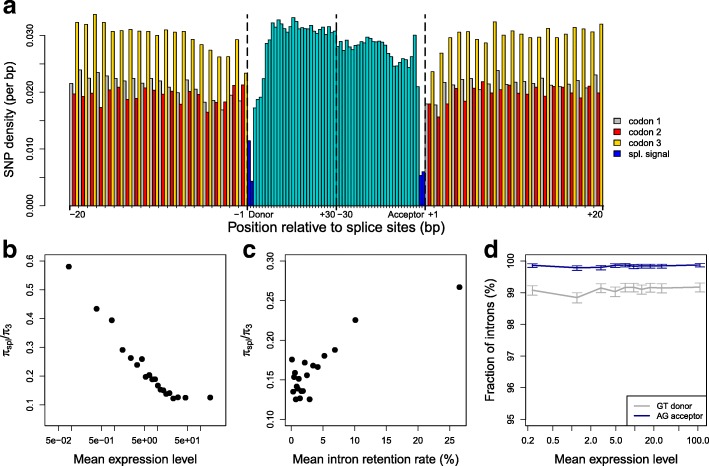
Results: We propose a test to quantify the fraction of AS events corresponding to errors. This test is based on the fact that the fitness cost of splicing errors increases with the number of introns in a gene and with expression level. We analyzed the transcriptome of the intron-rich eukaryote Paramecium tetraurelia. We show that in both normal and in NMD-deficient cells, AS rates strongly decrease with increasing expression level and with increasing number of introns. This relationship is observed for AS events that are detectable by NMD as well as for those that are not, which invalidates the hypothesis of a link with the regulation of gene expression. Our results show that in genes with a median expression level, 92-98% of observed splice variants correspond to errors. We observed the same patterns in human transcriptomes and we further show that AS rates correlate with the fitness cost of splicing errors.
Conclusions: These observations indicate that genes under weaker selective pressure accumulate more maladaptive substitutions and are more prone to splicing errors. Thus, to a large extent, patterns of gene expression variants simply reflect the balance between selection, mutation, and drift.
Keywords: Alternative splicing; Random genetic drift; Selectionist/neutralist debate.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
