

Beyond the hype: deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set
- PMID: 29086168
- PMCID: PMC5555960
- DOI: 10.1186/s13321-017-0232-0
Beyond the hype: deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set
Abstract
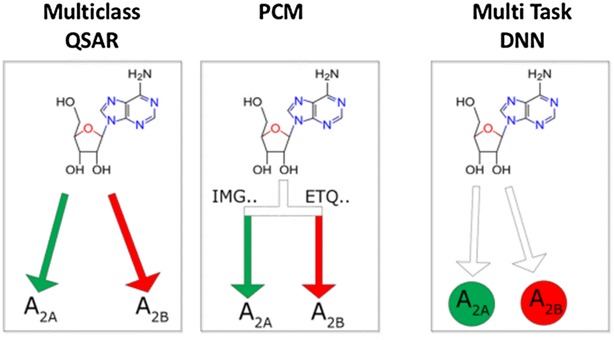
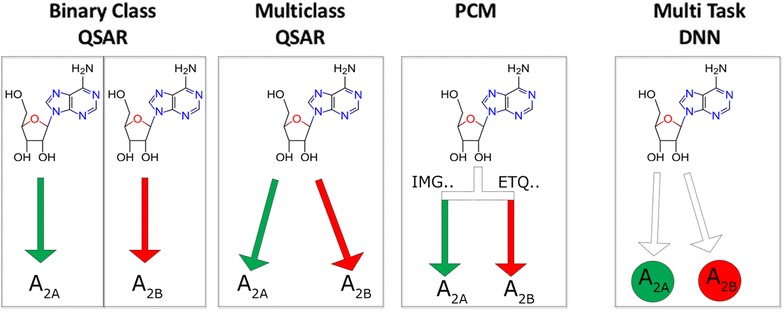
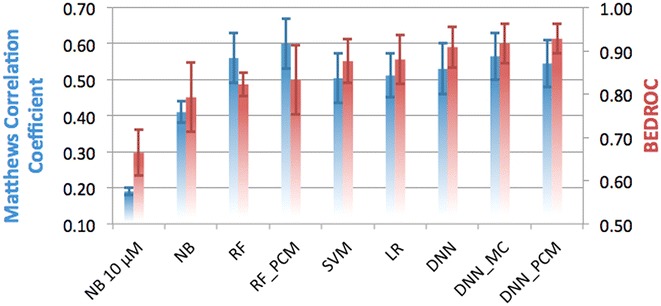
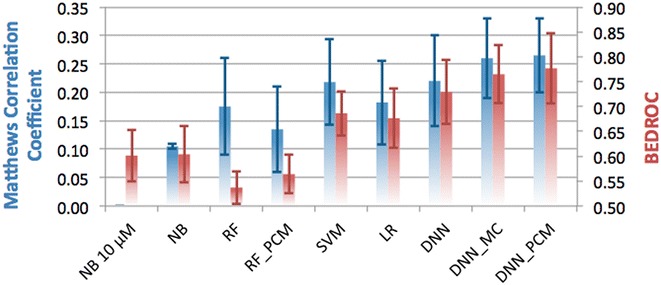
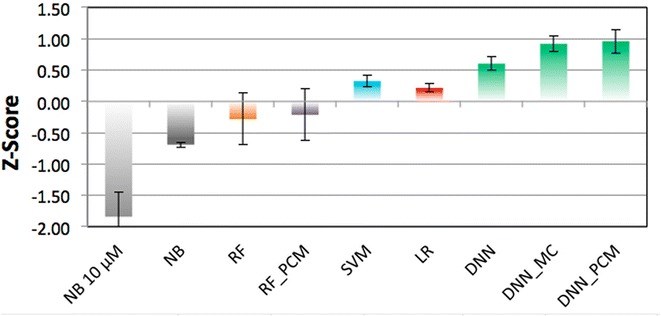
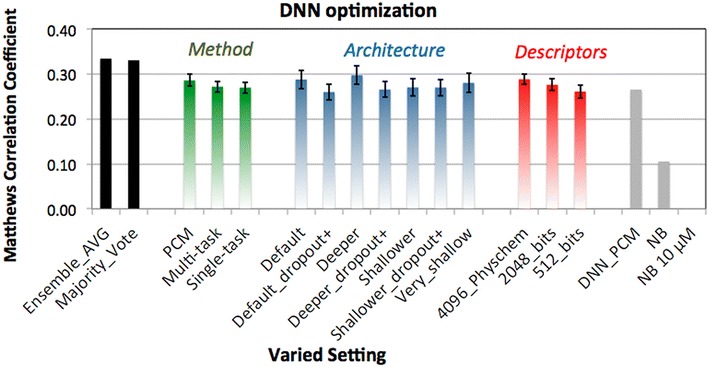
The increase of publicly available bioactivity data in recent years has fueled and catalyzed research in chemogenomics, data mining, and modeling approaches. As a direct result, over the past few years a multitude of different methods have been reported and evaluated, such as target fishing, nearest neighbor similarity-based methods, and Quantitative Structure Activity Relationship (QSAR)-based protocols. However, such studies are typically conducted on different datasets, using different validation strategies, and different metrics. In this study, different methods were compared using one single standardized dataset obtained from ChEMBL, which is made available to the public, using standardized metrics (BEDROC and Matthews Correlation Coefficient). Specifically, the performance of Naïve Bayes, Random Forests, Support Vector Machines, Logistic Regression, and Deep Neural Networks was assessed using QSAR and proteochemometric (PCM) methods. All methods were validated using both a random split validation and a temporal validation, with the latter being a more realistic benchmark of expected prospective execution. Deep Neural Networks are the top performing classifiers, highlighting the added value of Deep Neural Networks over other more conventional methods. Moreover, the best method ('DNN_PCM') performed significantly better at almost one standard deviation higher than the mean performance. Furthermore, Multi-task and PCM implementations were shown to improve performance over single task Deep Neural Networks. Conversely, target prediction performed almost two standard deviations under the mean performance. Random Forests, Support Vector Machines, and Logistic Regression performed around mean performance. Finally, using an ensemble of DNNs, alongside additional tuning, enhanced the relative performance by another 27% (compared with unoptimized 'DNN_PCM'). Here, a standardized set to test and evaluate different machine learning algorithms in the context of multi-task learning is offered by providing the data and the protocols. Graphical Abstract .
Keywords: ChEMBL; Cheminformatics; Chemogenomics; Deep neural networks; Proteochemometrics; QSAR.
Figures
References
-
- Protein Data Bank (2017) Yearly growth of total structures 2017 [July 7th 2017]. http://www.rcsb.org/pdb/statistics/contentGrowthChart.do?content=total
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
