

Choline transporter mutations in severe congenital myasthenic syndrome disrupt transporter localization
- PMID: 29088354
- PMCID: PMC5844214
- DOI: 10.1093/brain/awx249
Choline transporter mutations in severe congenital myasthenic syndrome disrupt transporter localization
Abstract
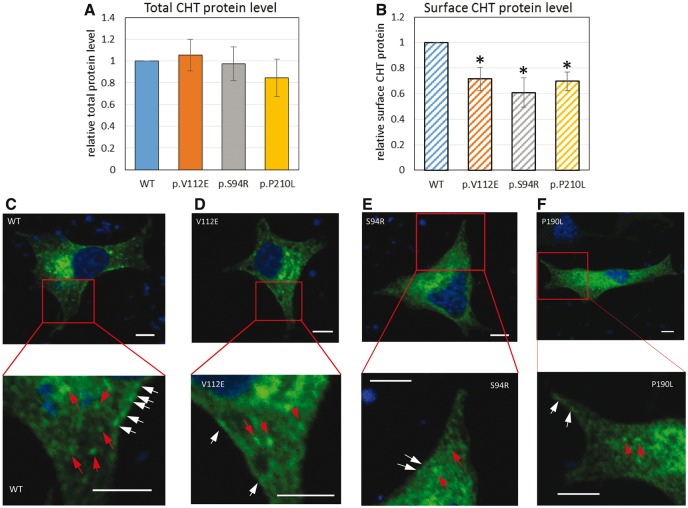
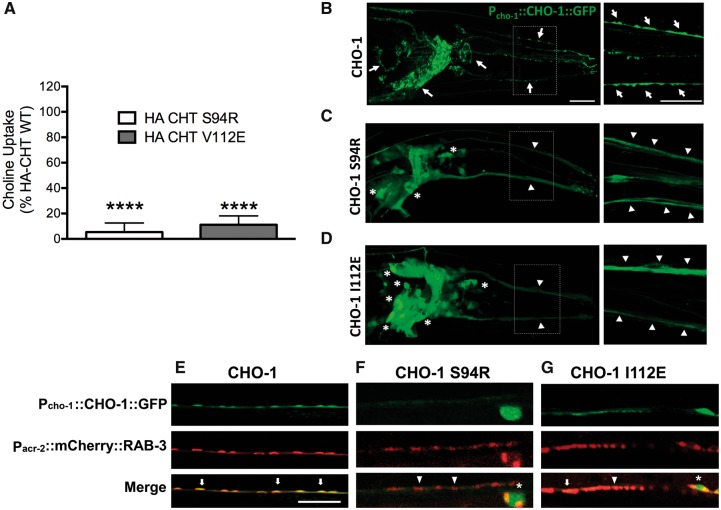
The presynaptic, high-affinity choline transporter is a critical determinant of signalling by the neurotransmitter acetylcholine at both central and peripheral cholinergic synapses, including the neuromuscular junction. Here we describe an autosomal recessive presynaptic congenital myasthenic syndrome presenting with a broad clinical phenotype due to homozygous choline transporter missense mutations. The clinical phenotype ranges from the classical presentation of a congenital myasthenic syndrome in one patient (p.Pro210Leu), to severe neurodevelopmental delay with brain atrophy (p.Ser94Arg) and extend the clinical outcomes to a more severe spectrum with infantile lethality (p.Val112Glu). Cells transfected with mutant transporter construct revealed a virtually complete loss of transport activity that was paralleled by a reduction in transporter cell surface expression. Consistent with these findings, studies to determine the impact of gene mutations on the trafficking of the Caenorhabditis elegans choline transporter orthologue revealed deficits in transporter export to axons and nerve terminals. These findings contrast with our previous findings in autosomal dominant distal hereditary motor neuropathy of a dominant-negative frameshift mutation at the C-terminus of choline transporter that was associated with significantly reduced, but not completely abrogated choline transporter function. Together our findings define divergent neuropathological outcomes arising from different classes of choline transporter mutation with distinct disease processes and modes of inheritance. These findings underscore the essential role played by the choline transporter in sustaining acetylcholine neurotransmission at both central and neuromuscular synapses, with important implications for treatment and drug selection.
Keywords: CHT; CHT trafficking; SLC5A7; choline uptake; congenital myasthenic syndrome.
© The Author (2017). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures

References
-
- Apparsundaram S, Ferguson SM, George AL Jr, Blakely RD. Molecular cloning of a human, hemicholinium-3-sensitive choline transporter. Biochem Biophys Res Commun 2000; 276: 862–7. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
