

Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition
- PMID: 29088702
- PMCID: PMC5933935
- DOI: 10.1038/nature24297
Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition
Abstract
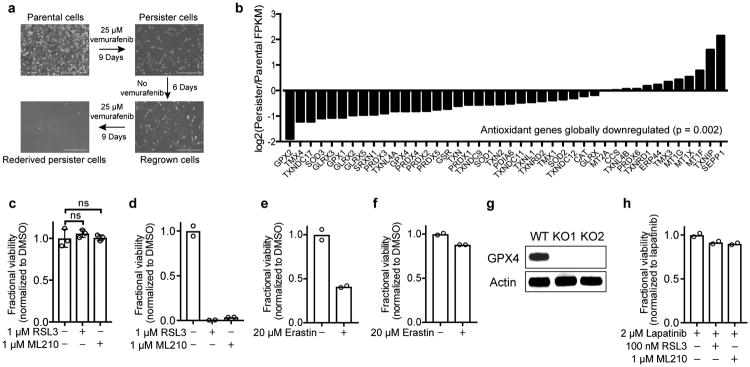
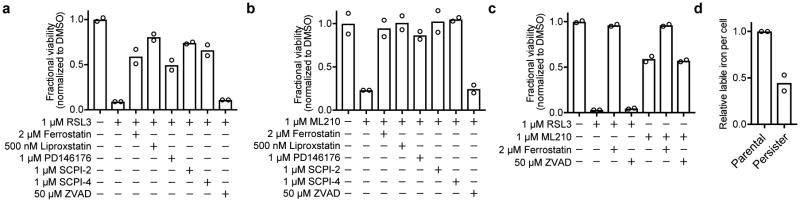
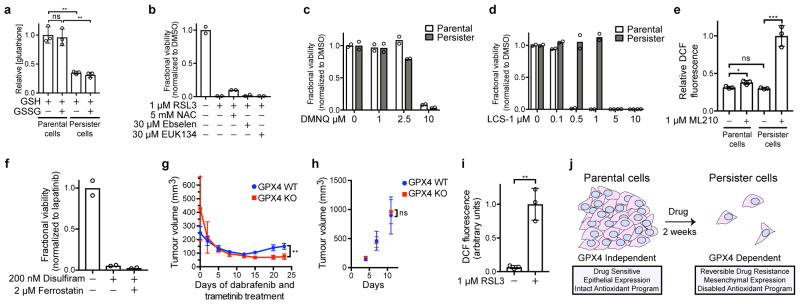
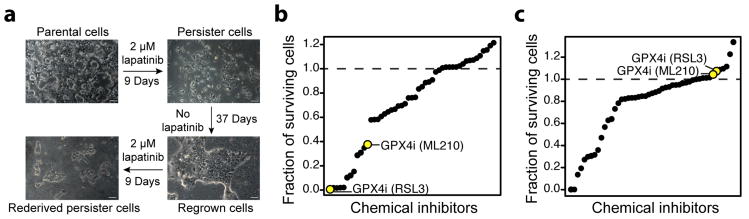
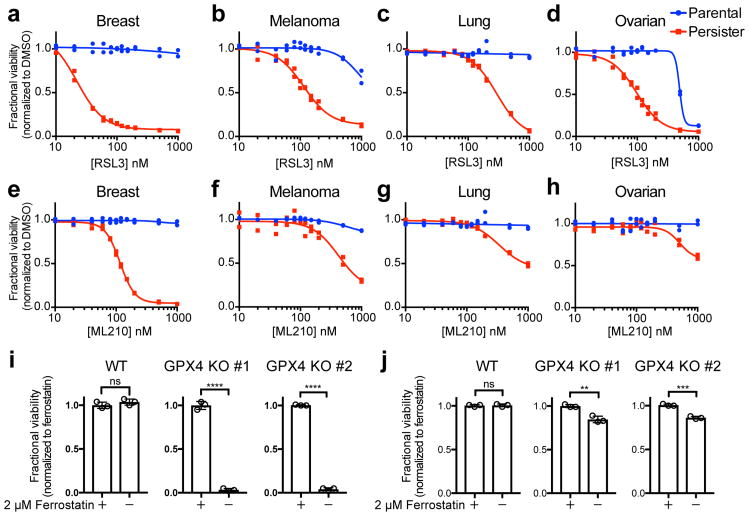
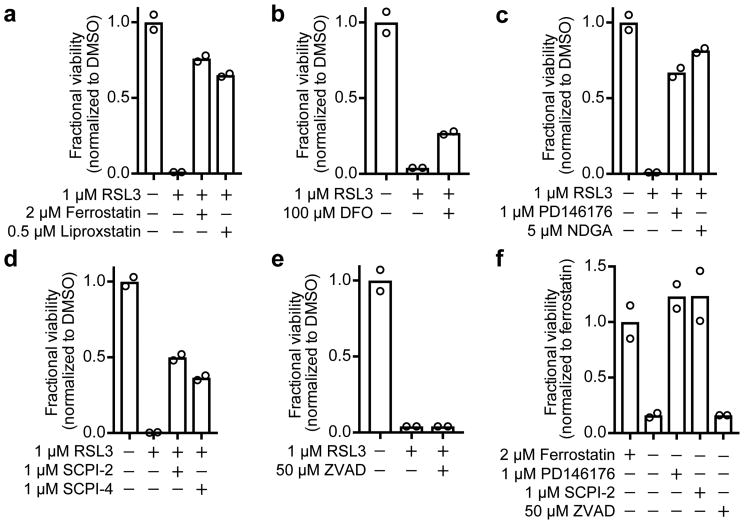
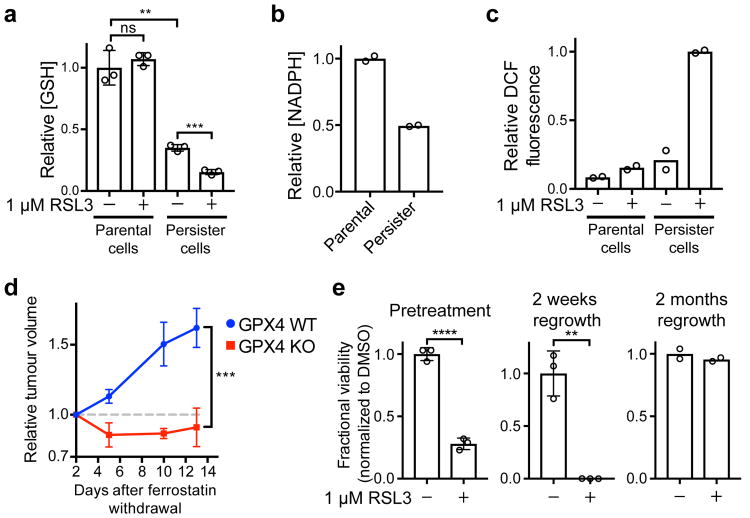
Acquired drug resistance prevents cancer therapies from achieving stable and complete responses. Emerging evidence implicates a key role for non-mutational drug resistance mechanisms underlying the survival of residual cancer 'persister' cells. The persister cell pool constitutes a reservoir from which drug-resistant tumours may emerge. Targeting persister cells therefore presents a therapeutic opportunity to impede tumour relapse. We previously found that cancer cells in a high mesenchymal therapy-resistant cell state are dependent on the lipid hydroperoxidase GPX4 for survival. Here we show that a similar therapy-resistant cell state underlies the behaviour of persister cells derived from a wide range of cancers and drug treatments. Consequently, we demonstrate that persister cells acquire a dependency on GPX4. Loss of GPX4 function results in selective persister cell ferroptotic death in vitro and prevents tumour relapse in mice. These findings suggest that targeting of GPX4 may represent a therapeutic strategy to prevent acquired drug resistance.
Figures
References
Additional references
-
- Salt MB, Bandyopadhyay S, McCormick F. Epithelial--to-mesenchymal transition rewires the molecular path to PI3K-dependent proliferation. Cancer Discov. 2014;4:186–199. doi: 10.1158/2159-8290.CD-13-0520. - DOI - PubMed
-
- Gelain DP, et al. A systematic review of human antioxidant genes. Front Biosci (Landmark Ed) 2009;14:4457–4463. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
