

A communal catalogue reveals Earth's multiscale microbial diversity
- PMID: 29088705
- PMCID: PMC6192678
- DOI: 10.1038/nature24621
A communal catalogue reveals Earth's multiscale microbial diversity
Abstract
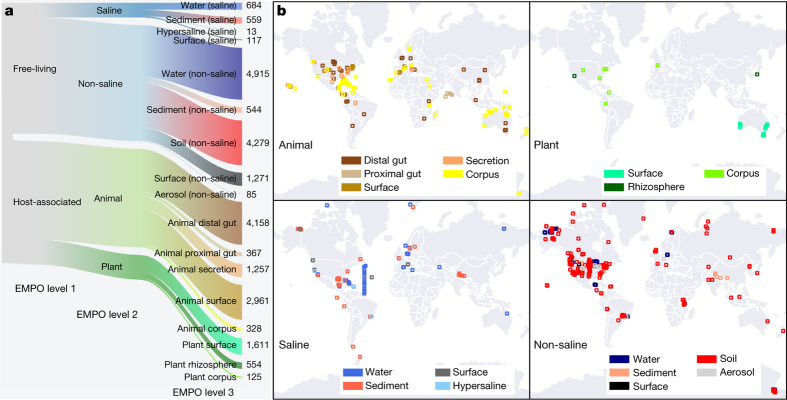
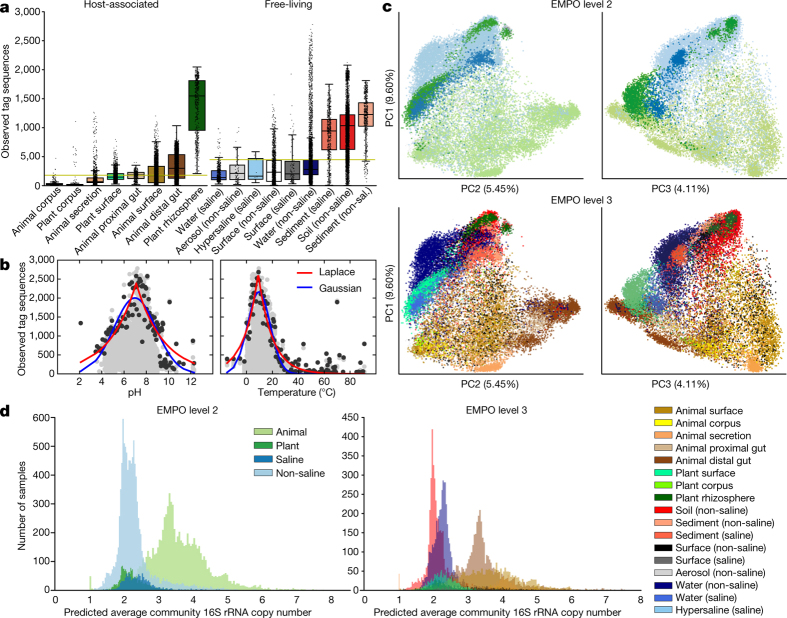
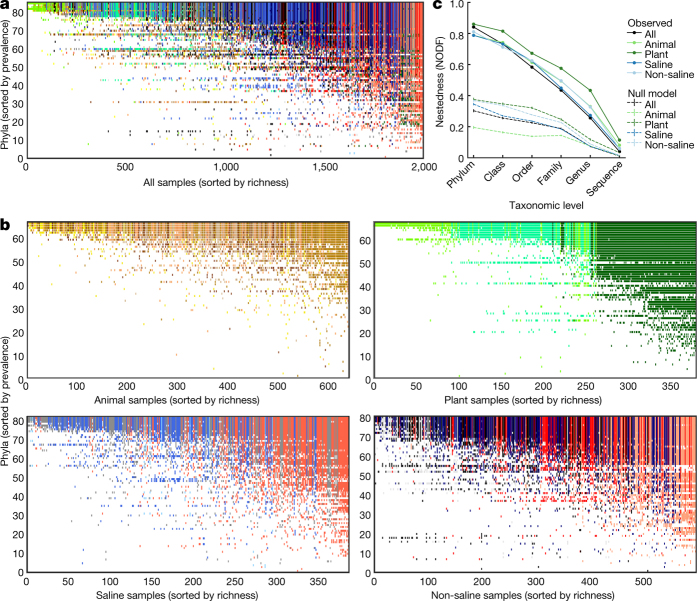
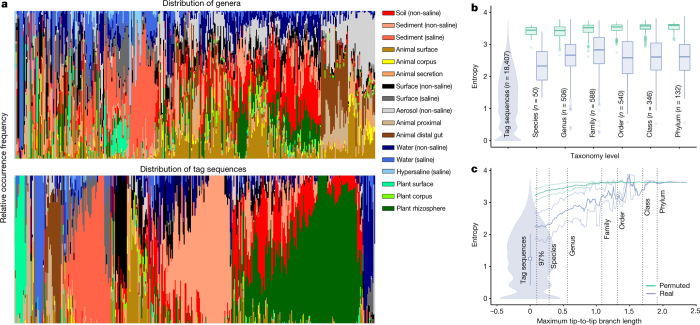
Our growing awareness of the microbial world's importance and diversity contrasts starkly with our limited understanding of its fundamental structure. Despite recent advances in DNA sequencing, a lack of standardized protocols and common analytical frameworks impedes comparisons among studies, hindering the development of global inferences about microbial life on Earth. Here we present a meta-analysis of microbial community samples collected by hundreds of researchers for the Earth Microbiome Project. Coordinated protocols and new analytical methods, particularly the use of exact sequences instead of clustered operational taxonomic units, enable bacterial and archaeal ribosomal RNA gene sequences to be followed across multiple studies and allow us to explore patterns of diversity at an unprecedented scale. The result is both a reference database giving global context to DNA sequence data and a framework for incorporating data from future studies, fostering increasingly complete characterization of Earth's microbial diversity.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
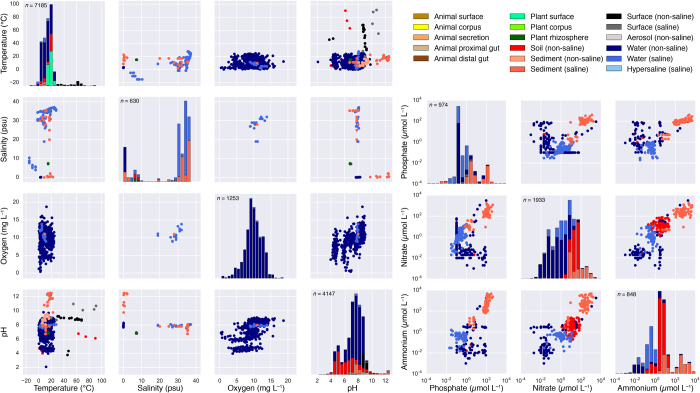
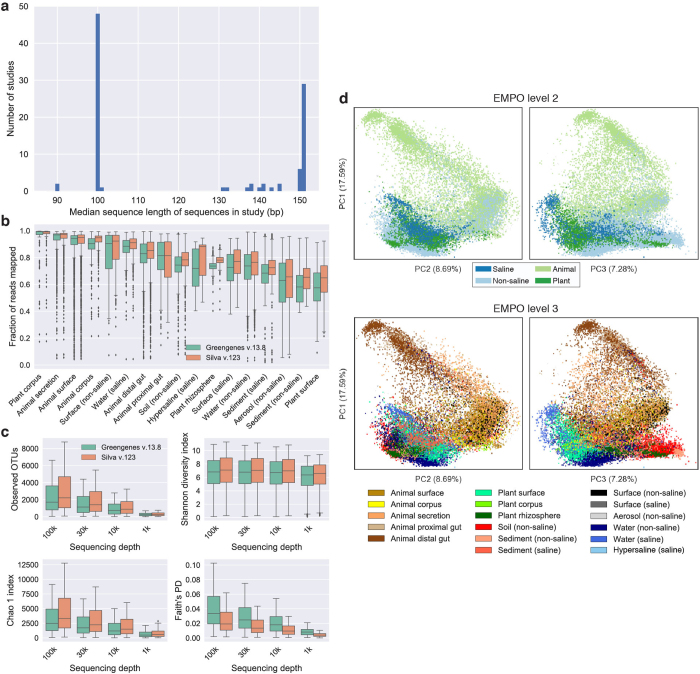
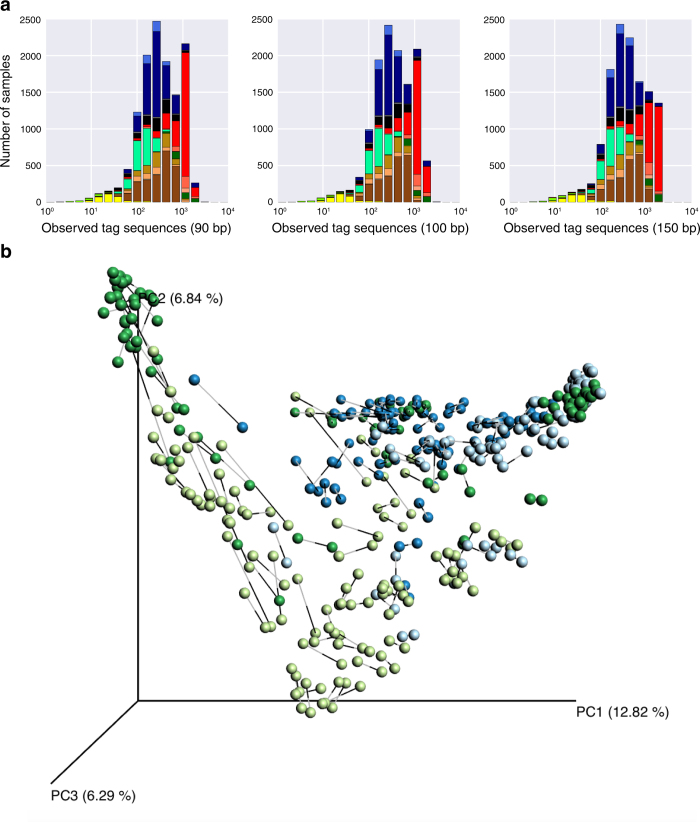
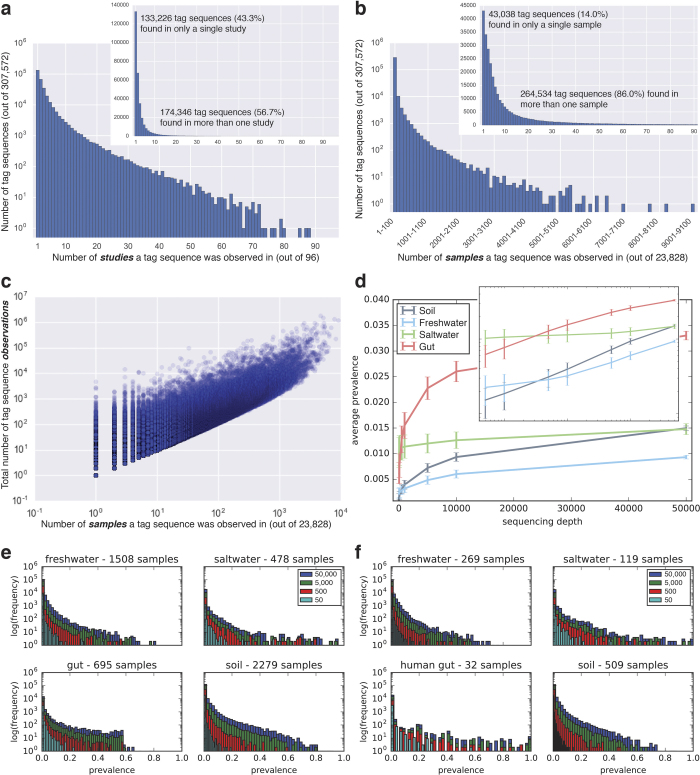
References
-
- Philippot L, Raaijmakers JM, Lemanceau P, van der Putten WH. Going back to the roots: the microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013;11:789–799. - PubMed
