

Chasing the open-state structure of pentameric ligand-gated ion channels
- PMID: 29089419
- PMCID: PMC5715906
- DOI: 10.1085/jgp.201711803
Chasing the open-state structure of pentameric ligand-gated ion channels
Abstract
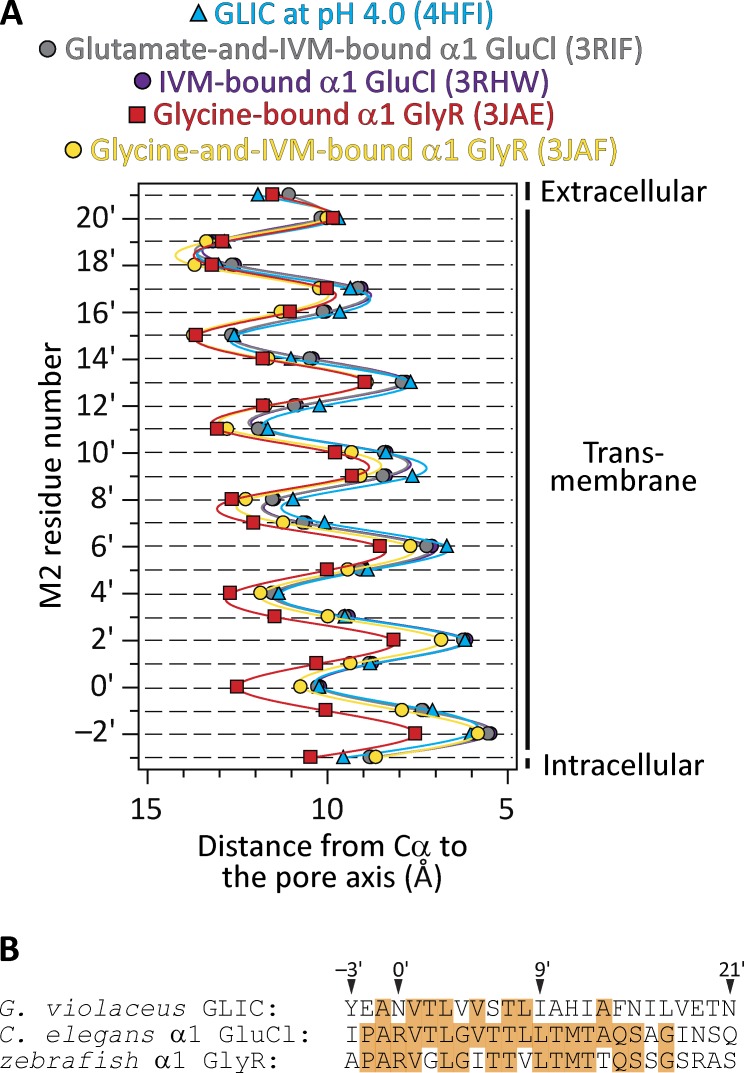
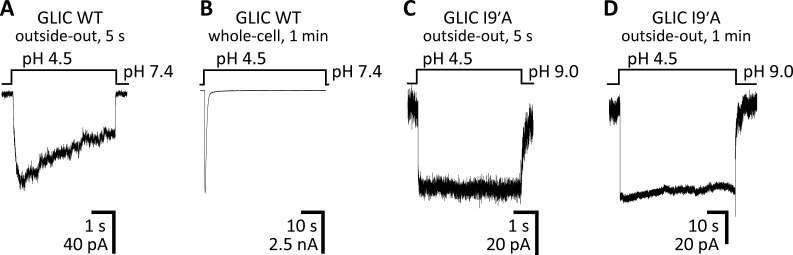
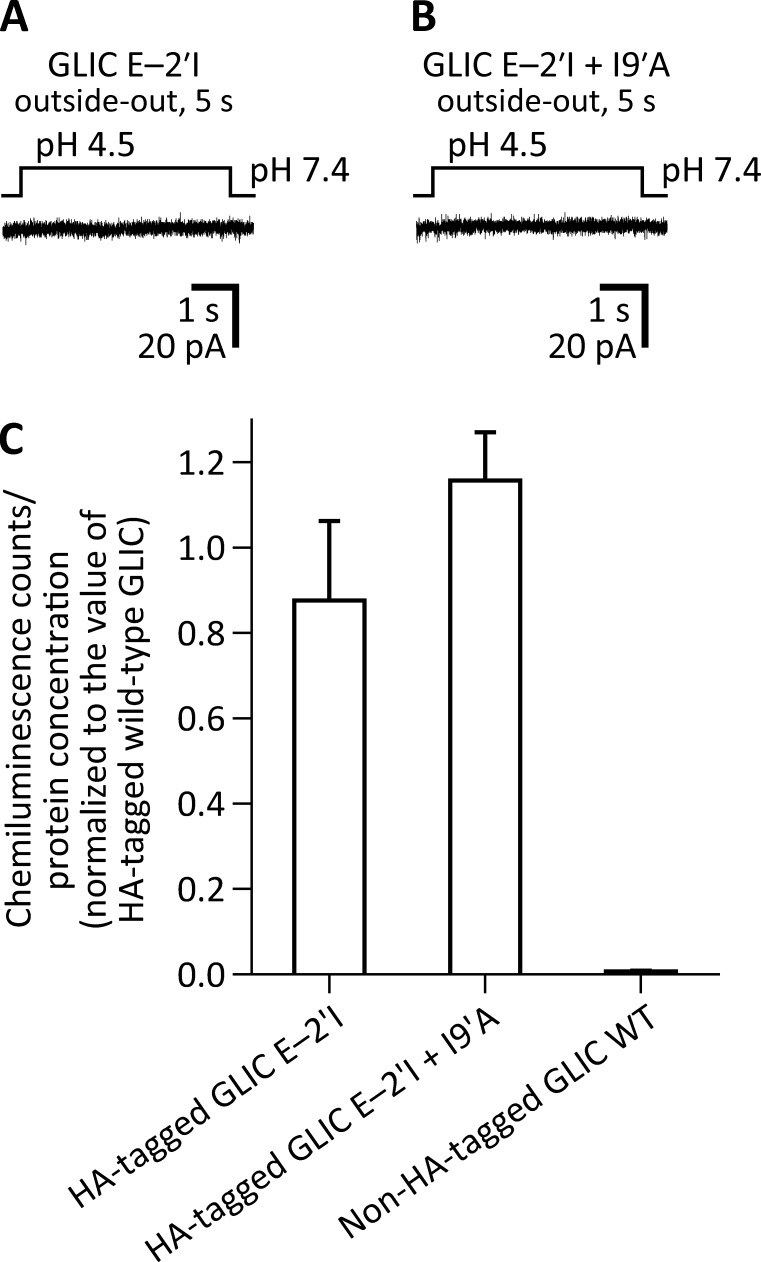
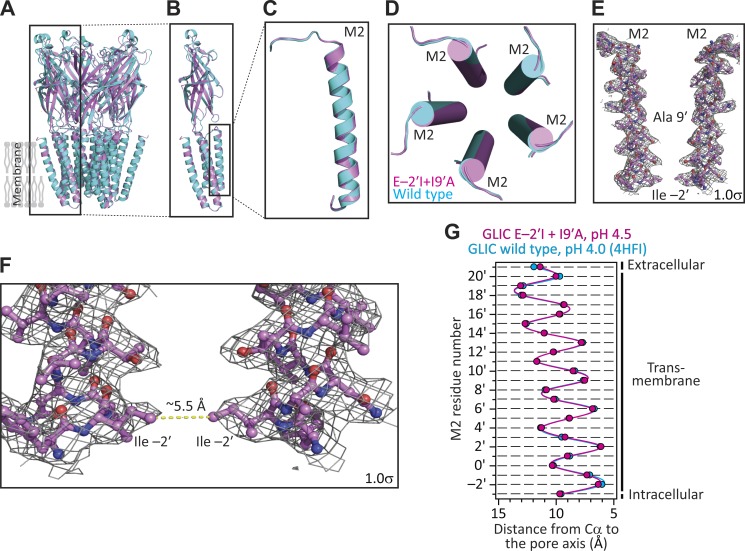
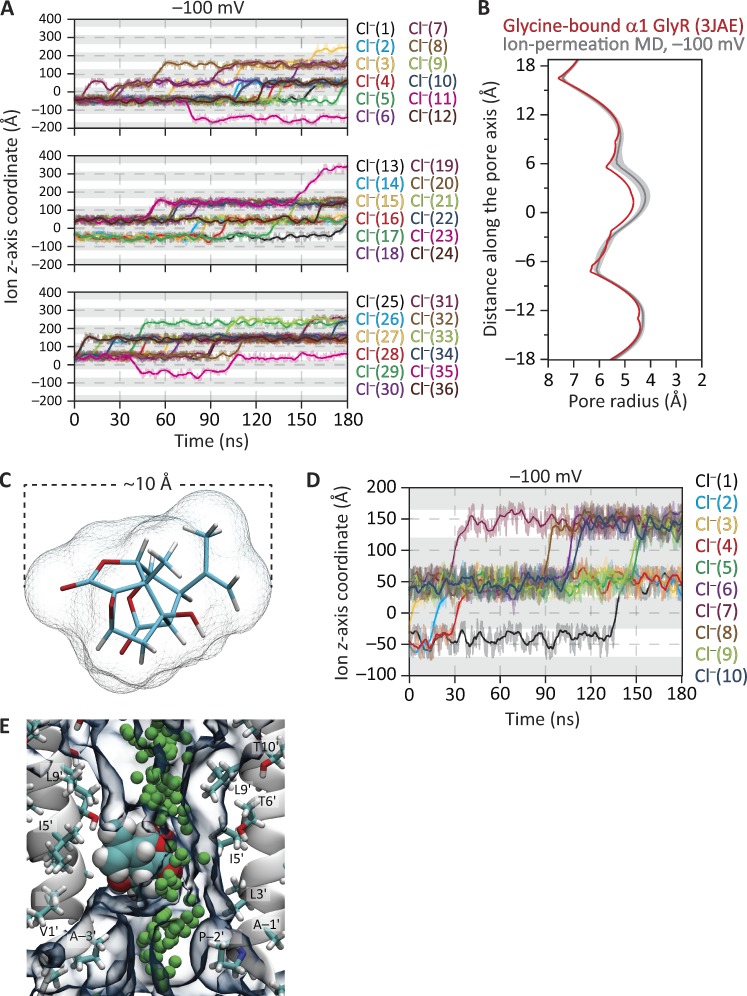
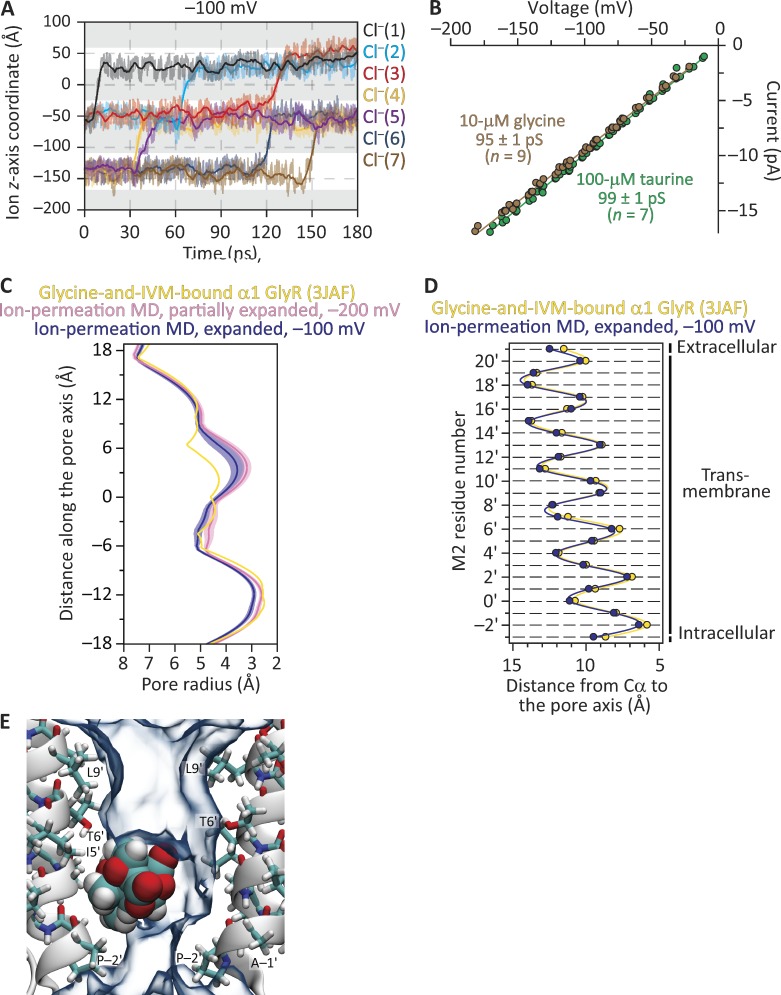
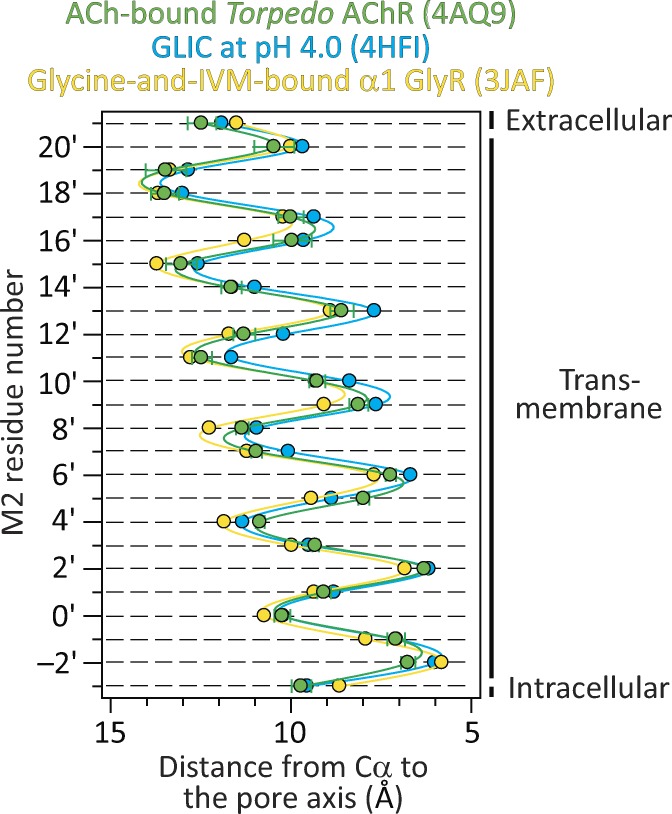
Remarkable advances have been made toward the structural characterization of ion channels in the last two decades. However, the unambiguous assignment of well-defined functional states to the obtained structural models has proved challenging. In the case of the superfamily of nicotinic-receptor channels (also referred to as pentameric ligand-gated ion channels [pLGICs]), for example, two different types of model of the open-channel conformation have been proposed on the basis of structures solved to resolutions better than 4.0 Å. At the level of the transmembrane pore, the open-state models of the proton-gated pLGIC from Gloeobacter violaceus (GLIC) and the invertebrate glutamate-gated Cl- channel (GluCl) are very similar to each other, but that of the glycine receptor (GlyR) is considerably wider. Indeed, the mean distances between the axis of ion permeation and the Cα atoms at the narrowest constriction of the pore (position -2') differ by ∼2 Å in these two classes of model, a large difference when it comes to understanding the physicochemical bases of ion conduction and charge selectivity. Here, we take advantage of the extreme open-channel stabilizing effect of mutations at pore-facing position 9'. We find that the I9'A mutation slows down entry into desensitization of GLIC to the extent that macroscopic currents decay only slightly by the end of pH 4.5 solution applications to the extracellular side for several minutes. We crystallize (at pH 4.5) two variants of GLIC carrying this mutation and solve their structures to resolutions of 3.12 Å and 3.36 Å. Furthermore, we perform all-atom molecular dynamics simulations of ion permeation and picrotoxinin block, using the different open-channel structural models. On the basis of these results, we favor the notion that the open-channel structure of pLGICs from animals is much closer to that of the narrow models (of GLIC and GluCl) than it is to that of the GlyR.
© 2017 Gonzalez-Gutierrez et al.
Figures
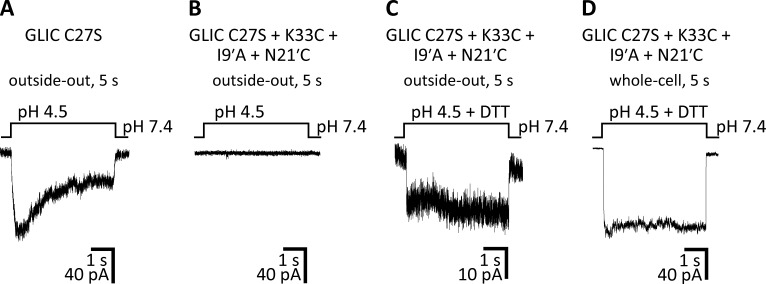
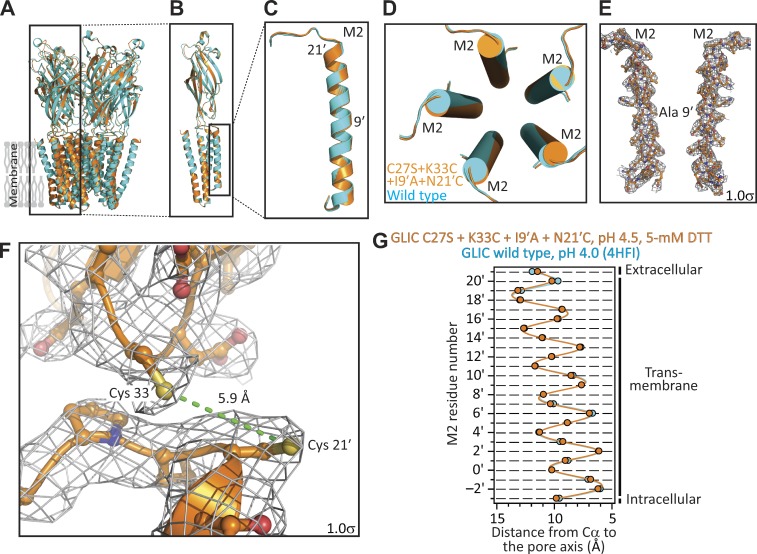
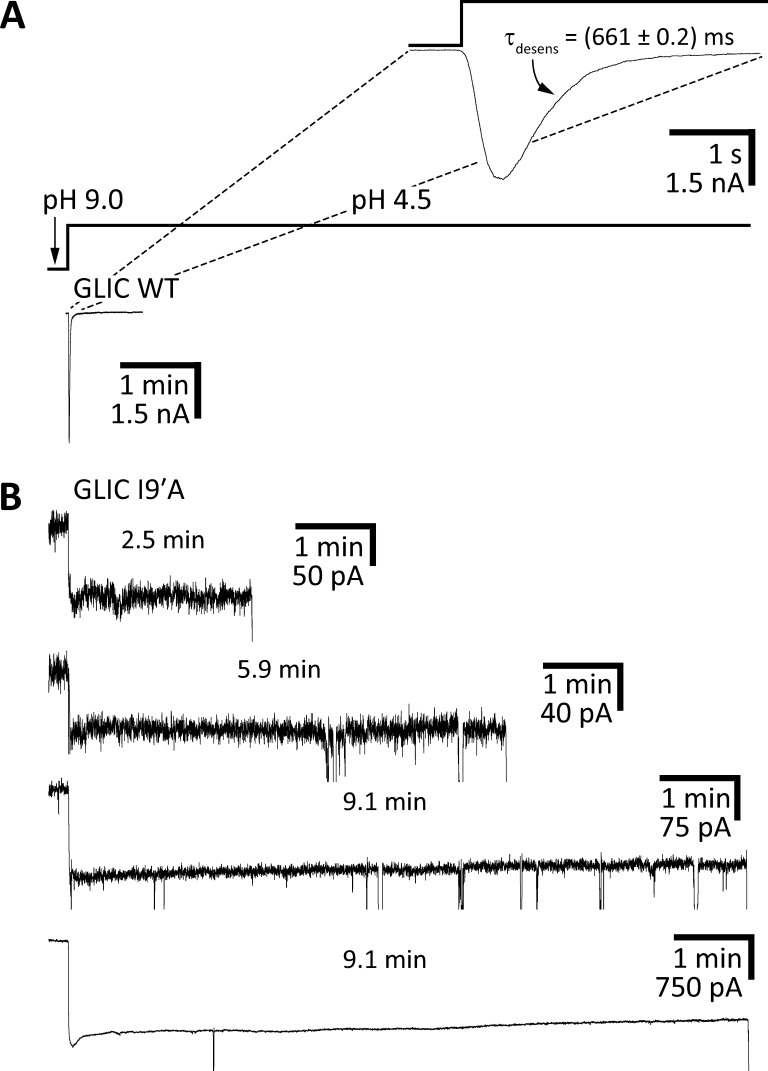
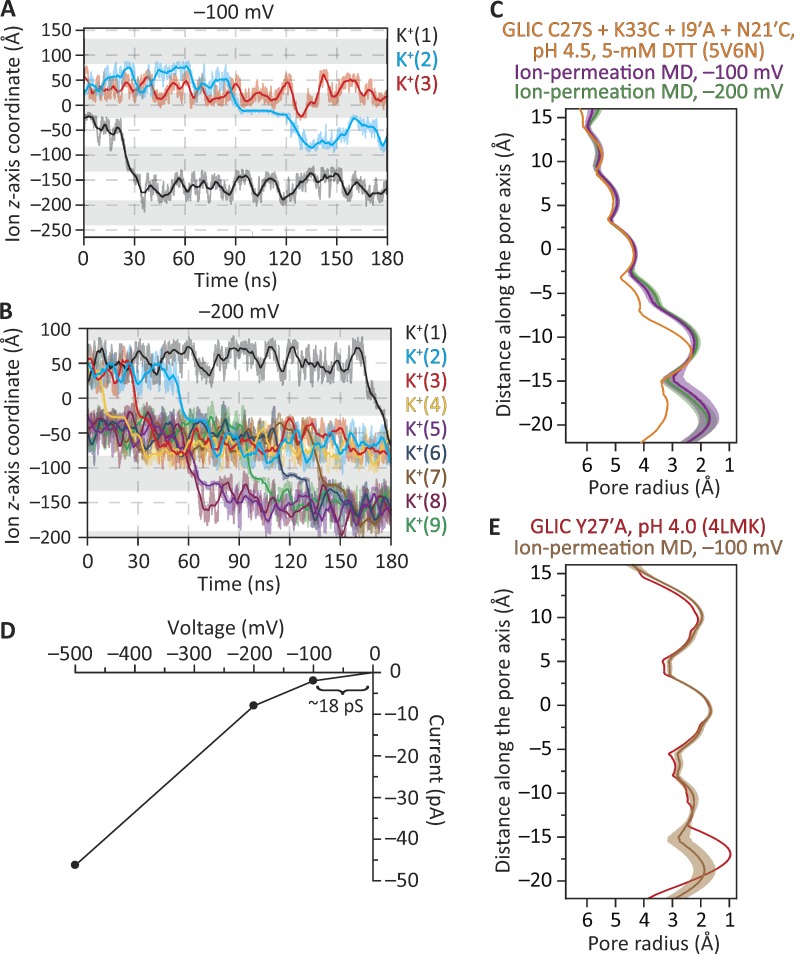
References
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
