

Pan-cancer analysis of homozygous deletions in primary tumours uncovers rare tumour suppressors
- PMID: 29089486
- PMCID: PMC5663922
- DOI: 10.1038/s41467-017-01355-0
Pan-cancer analysis of homozygous deletions in primary tumours uncovers rare tumour suppressors
Erratum in
-
Author Correction: Pan-cancer analysis of homozygous deletions in primary tumours uncovers rare tumour suppressors.Nat Commun. 2018 Dec 17;9(1):5397. doi: 10.1038/s41467-018-07842-2. Nat Commun. 2018. PMID: 30559362 Free PMC article.
-
Author Correction: Pan-cancer analysis of homozygous deletions in primary tumours uncovers rare tumour suppressors.Nat Commun. 2019 Jan 28;10(1):525. doi: 10.1038/s41467-019-08512-7. Nat Commun. 2019. PMID: 30692535 Free PMC article.
Abstract
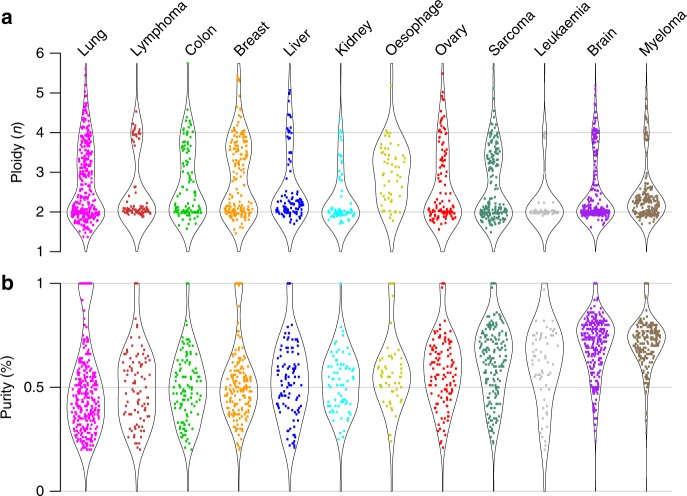
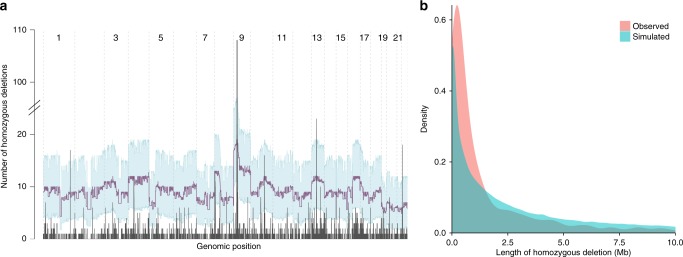
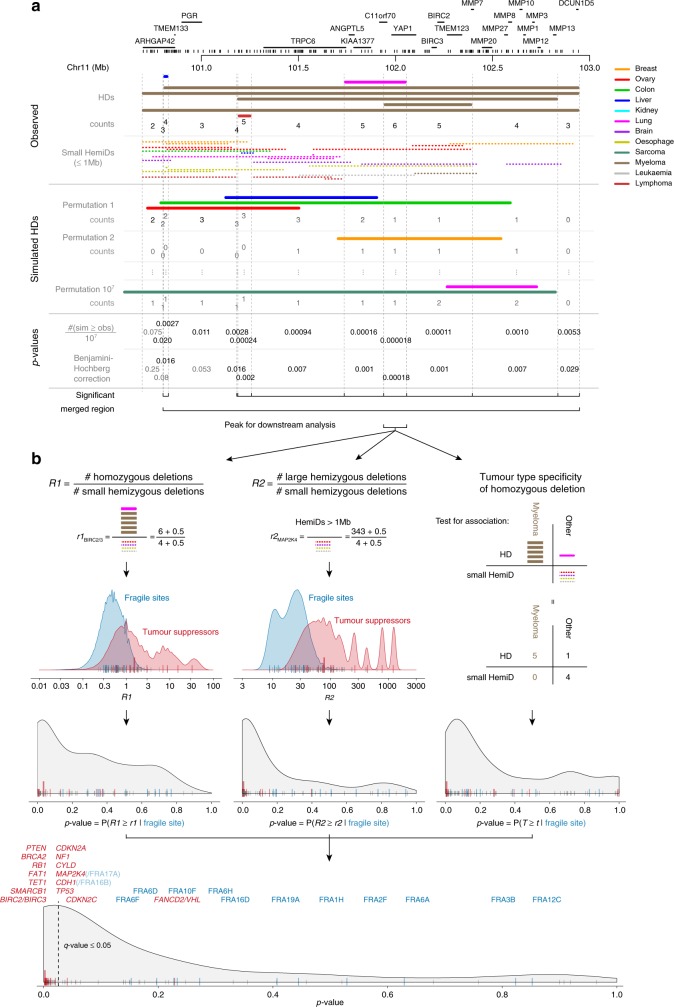
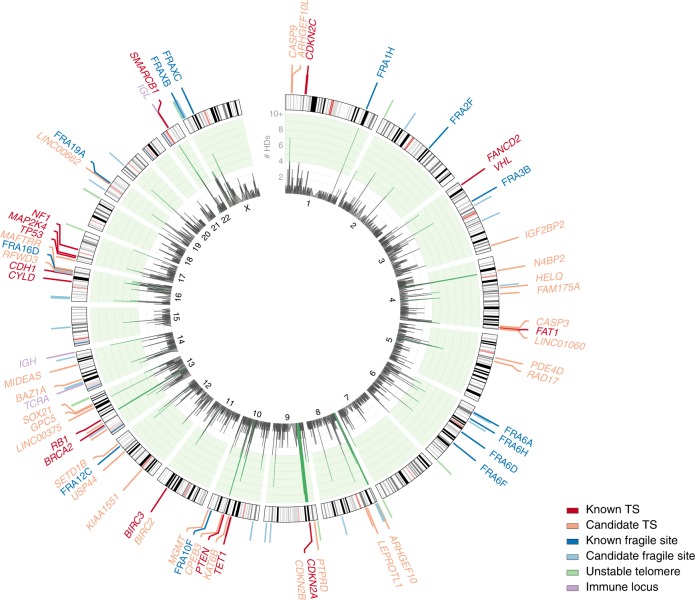
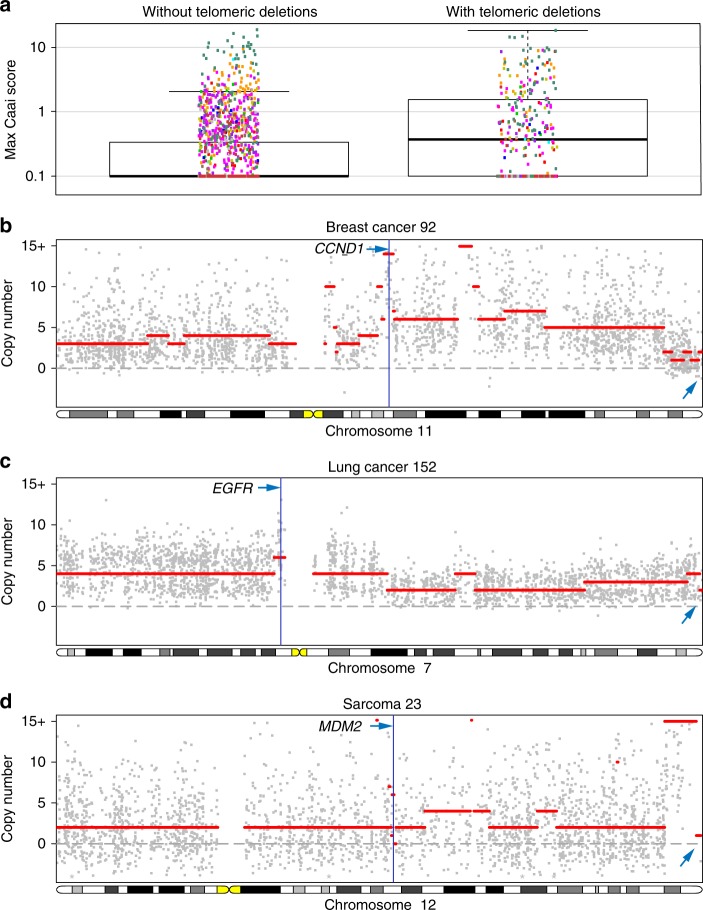
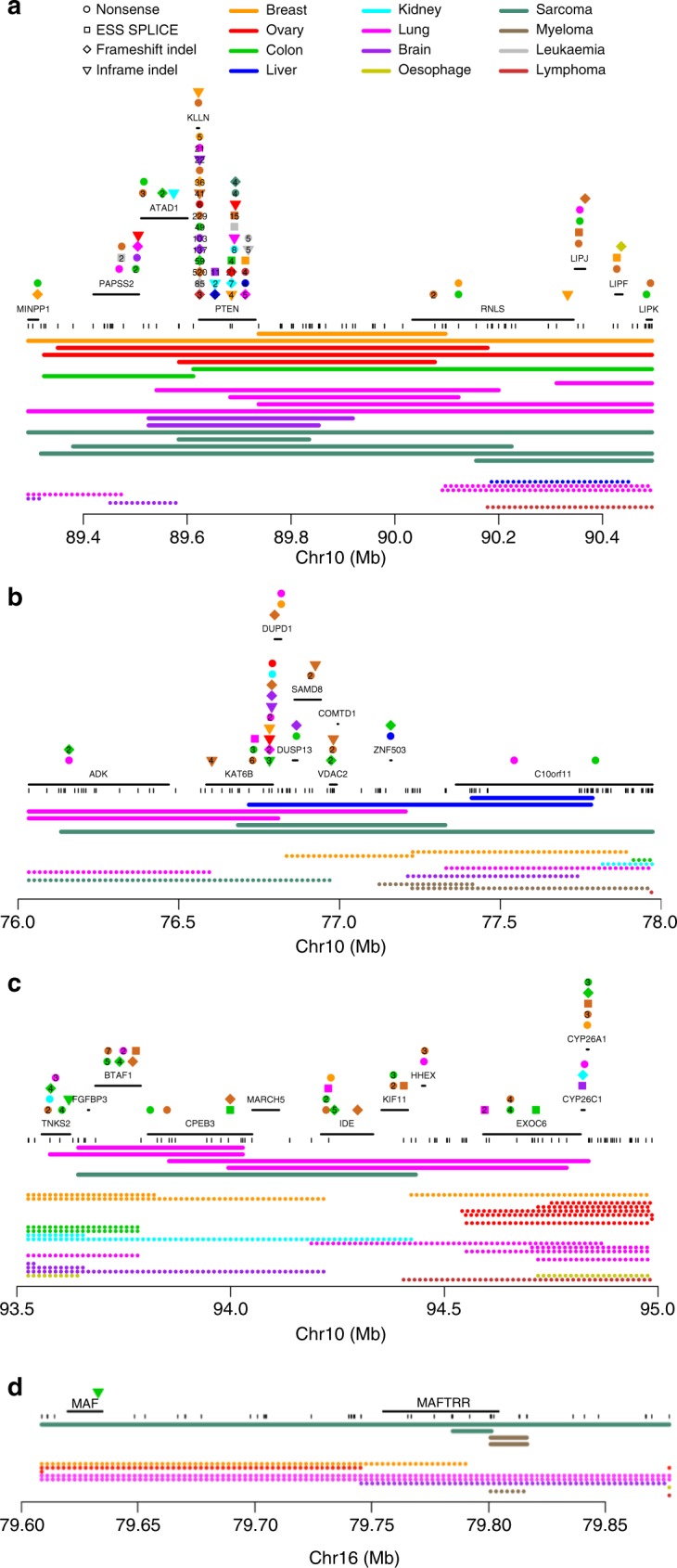
Homozygous deletions are rare in cancers and often target tumour suppressor genes. Here, we build a compendium of 2218 primary tumours across 12 human cancer types and systematically screen for homozygous deletions, aiming to identify rare tumour suppressors. Our analysis defines 96 genomic regions recurrently targeted by homozygous deletions. These recurrent homozygous deletions occur either over tumour suppressors or over fragile sites, regions of increased genomic instability. We construct a statistical model that separates fragile sites from regions showing signatures of positive selection for homozygous deletions and identify candidate tumour suppressors within those regions. We find 16 established tumour suppressors and propose 27 candidate tumour suppressors. Several of these genes (including MGMT, RAD17, and USP44) show prior evidence of a tumour suppressive function. Other candidate tumour suppressors, such as MAFTRR, KIAA1551, and IGF2BP2, are novel. Our study demonstrates how rare tumour suppressors can be identified through copy number meta-analysis.
Conflict of interest statement
K.P.W. is President of Tempus Lab, Inc., Chicago, IL, USA.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
