

RNA sequencing and proteomics approaches reveal novel deficits in the cortex of Mecp2-deficient mice, a model for Rett syndrome
- PMID: 29090078
- PMCID: PMC5655833
- DOI: 10.1186/s13229-017-0174-4
RNA sequencing and proteomics approaches reveal novel deficits in the cortex of Mecp2-deficient mice, a model for Rett syndrome
Abstract
Background: Rett syndrome (RTT) is an X-linked neurodevelopmental disorder caused by mutations in the transcriptional regulator MeCP2. Much of our understanding of MeCP2 function is derived from transcriptomic studies with the general assumption that alterations in the transcriptome correlate with proteomic changes. Advances in mass spectrometry-based proteomics have facilitated recent interest in the examination of global protein expression to better understand the biology between transcriptional and translational regulation.
Methods: We therefore performed the first comprehensive transcriptome-proteome comparison in a RTT mouse model to elucidate RTT pathophysiology, identify potential therapeutic targets, and further our understanding of MeCP2 function. The whole cortex of wild-type and symptomatic RTT male littermates (n = 4 per genotype) were analyzed using RNA-sequencing and data-independent acquisition liquid chromatography tandem mass spectrometry. Ingenuity® Pathway Analysis was used to identify significantly affected pathways in the transcriptomic and proteomic data sets.
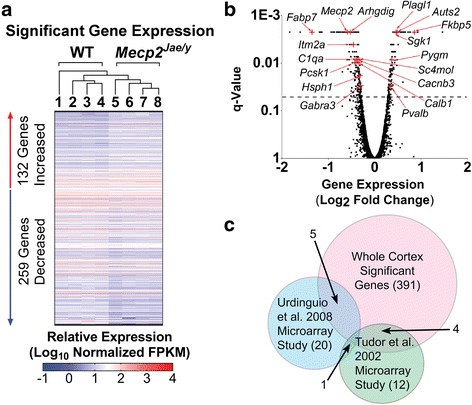
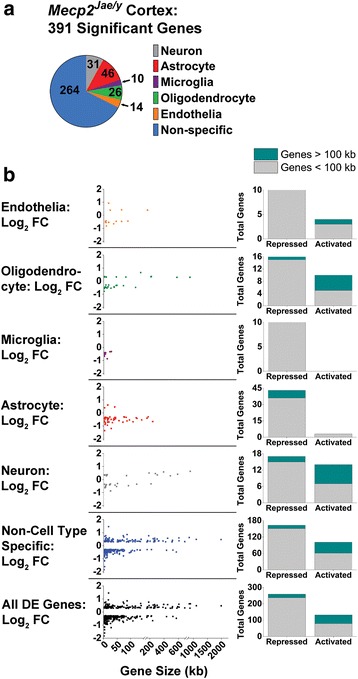
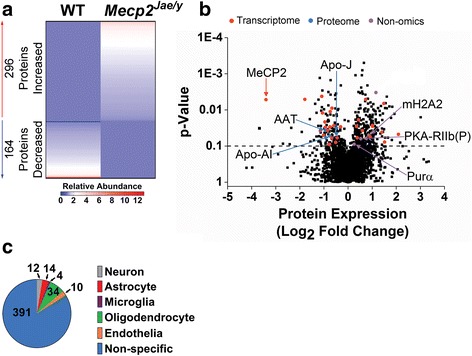
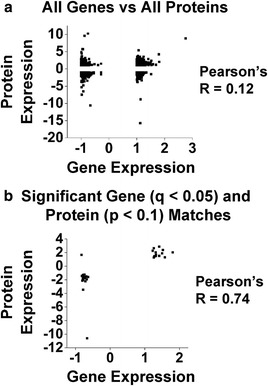
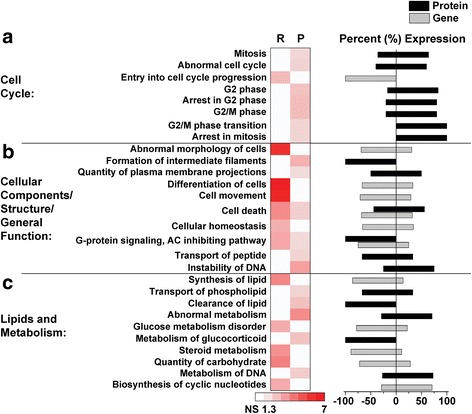
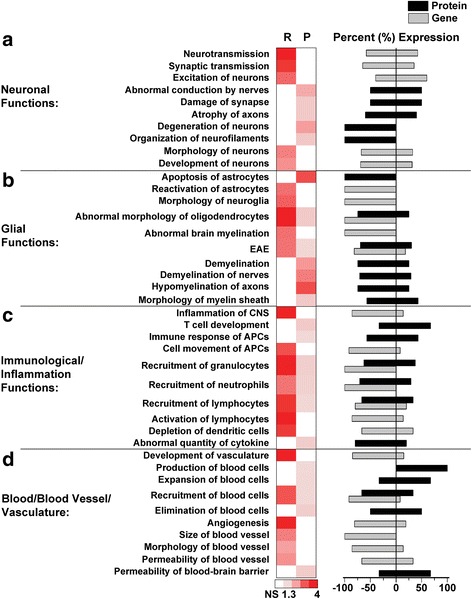
Results: Our results indicate these two "omics" data sets supplement one another. In addition to confirming previous works regarding mRNA expression in Mecp2-deficient animals, the current study identified hundreds of novel protein targets. Several selected protein targets were validated by Western blot analysis. These data indicate RNA metabolism, proteostasis, monoamine metabolism, and cholesterol synthesis are disrupted in the RTT proteome. Hits common to both data sets indicate disrupted cellular metabolism, calcium signaling, protein stability, DNA binding, and cytoskeletal cell structure. Finally, in addition to confirming disrupted pathways and identifying novel hits in neuronal structure and synaptic transmission, our data indicate aberrant myelination, inflammation, and vascular disruption. Intriguingly, there is no evidence of reactive gliosis, but instead, gene, protein, and pathway analysis suggest astrocytic maturation and morphological deficits.
Conclusions: This comparative omics analysis supports previous works indicating widespread CNS dysfunction and may serve as a valuable resource for those interested in cellular dysfunction in RTT.
Keywords: Multi-cellular deficits; Proteome; Rett syndrome; Transcriptome.
Conflict of interest statement
Ethics approval
All mouse experimental protocols were followed according to NIH guidelines and approval from the Animal Care and Use Committee of the University of Alabama at Birmingham.
Consent for publication
Not applicable.
Competing interests
MRH owns shares in Vulcan Analytical, LLC. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Cuddapah VA, Pillai RB, Shekar KV, Lane JB, Motil KJ, Skinner SA, Tarquinio DC, Glaze DG, McGwin G, Kaufmann WE, et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet. 2014;51:152–158. doi: 10.1136/jmedgenet-2013-102113. - DOI - PMC - PubMed
-
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
