

Prospective investigation of FOXP1 syndrome
- PMID: 29090079
- PMCID: PMC5655854
- DOI: 10.1186/s13229-017-0172-6
Prospective investigation of FOXP1 syndrome
Abstract
Background: Haploinsufficiency of the forkhead-box protein P1 (FOXP1) gene leads to a neurodevelopmental disorder termed FOXP1 syndrome. Previous studies in individuals carrying FOXP1 mutations and deletions have described the presence of autism spectrum disorder (ASD) traits, intellectual disability, language impairment, and psychiatric features. The goal of the present study was to comprehensively characterize the genetic and clinical spectrum of FOXP1 syndrome. This is the first study to prospectively examine the genotype-phenotype relationship in multiple individuals with FOXP1 syndrome, using a battery of standardized clinical assessments.
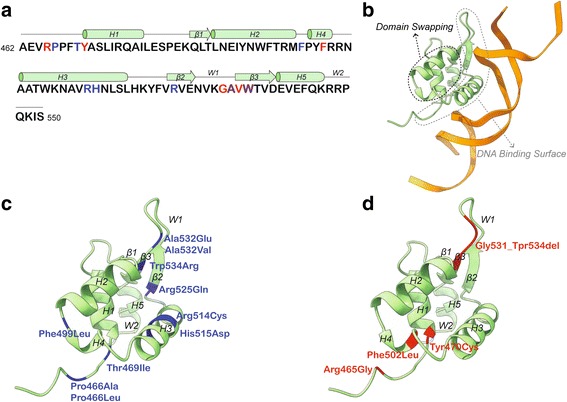
Methods: Genetic and clinical data was obtained and analyzed from nine children and adolescents between the ages of 5-17 with mutations in FOXP1. Phenotypic characterization included gold standard ASD testing and norm-referenced measures of cognition, adaptive behavior, language, motor, and visual-motor integration skills. In addition, psychiatric, medical, neurological, and dysmorphology examinations were completed by a multidisciplinary team of clinicians. A comprehensive review of reported cases was also performed. All missense and in-frame mutations were mapped onto the three-dimensional structure of DNA-bound FOXP1.
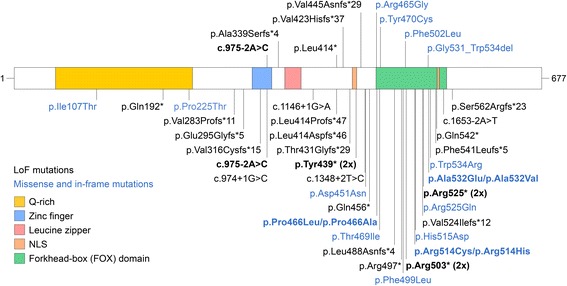
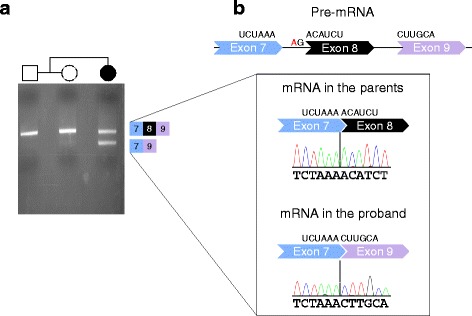
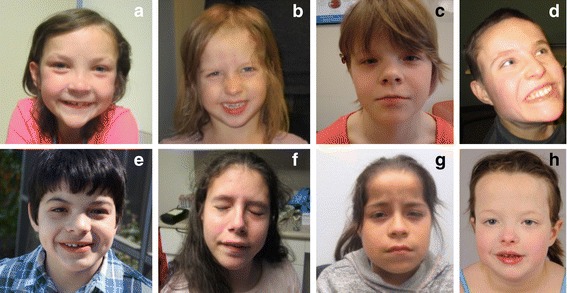
Results: We have identified nine de novo mutations, including three frameshift, one nonsense, one mutation in an essential splice site resulting in frameshift and insertion of a premature stop codon, three missense, and one in-frame deletion. Reviewing prior literature, we found seven instances of recurrent mutations and another 34 private mutations. The majority of pathogenic missense and in-frame mutations, including all four missense mutations in our cohort, lie in the DNA-binding domain. Through structural analyses, we show that the mutations perturb amino acids necessary for binding to the DNA or interfere with the domain swapping that mediates FOXP1 dimerization. Individuals with FOXP1 syndrome presented with delays in early motor and language milestones, language impairment (expressive language > receptive language), ASD symptoms, visual-motor integration deficits, and complex psychiatric presentations characterized by anxiety, obsessive-compulsive traits, attention deficits, and externalizing symptoms. Medical features included non-specific structural brain abnormalities and dysmorphic features, endocrine and gastrointestinal problems, sleep disturbances, and sinopulmonary infections.
Conclusions: This study identifies novel FOXP1 mutations associated with FOXP1 syndrome, identifies recurrent mutations, and demonstrates significant clustering of missense mutations in the DNA-binding domain. Clinical findings confirm the role FOXP1 plays in development across multiple domains of functioning. The genetic findings can be incorporated into clinical genetics practice to improve accurate genetic diagnosis of FOXP1 syndrome and the clinical findings can inform monitoring and treatment of individuals with FOXP1 syndrome.
Conflict of interest statement
Ethics approval and consent to participate
The study was approved by the Institutional Review Boards at the Icahn School of Medicine at Mount Sinai and at the University of Washington. Parents or legal guardians provided informed consent for participation.
Consent for publication
Written informed consent was obtained from parents or legal guardians of participants for publication of participants’ individuals details and accompanying images in this manuscript. The consent forms are held by the authors’ institutions in the patients’ clinical notes and are available for review by the Editor-in-Chief.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M, Foomani G, Dobrzeniecka S, Krebs MO, Joober R, et al. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet. 2010;87:671–678. doi: 10.1016/j.ajhg.2010.09.017. - DOI - PMC - PubMed
-
- Horn D, Kapeller J, Rivera-Brugues N, Moog U, Lorenz-Depiereux B, Eck S, Hempel M, Wagenstaller J, Gawthrope A, Monaco AP, et al. Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum Mutat. 2010;31:E1851–E1860. doi: 10.1002/humu.21362. - DOI - PMC - PubMed
-
- Sollis E, Graham SA, Vino A, Froehlich H, Vreeburg M, Dimitropoulou D, Gilissen C, Pfundt R, Rappold GA, Brunner HG, et al. Identification and functional characterization of de novo FOXP1 variants provides novel insights into the etiology of neurodevelopmental disorder. Hum Mol Genet. 2016;25:546–557. doi: 10.1093/hmg/ddv495. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
