

Demographics and patient characteristics of 1209 patients with Gaucher disease: Descriptive analysis from the Gaucher Outcome Survey (GOS)
- PMID: 29090476
- PMCID: PMC5814927
- DOI: 10.1002/ajh.24957
Demographics and patient characteristics of 1209 patients with Gaucher disease: Descriptive analysis from the Gaucher Outcome Survey (GOS)
Abstract
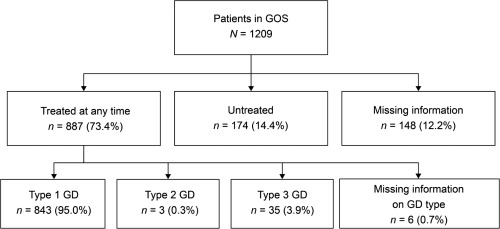
The Gaucher Outcome Survey (GOS) is an international Gaucher disease (GD) registry established in 2010 for patients with a confirmed GD diagnosis, regardless of GD type or treatment status, designed to evaluate the safety and long-term effectiveness of velaglucerase alfa and other GD-related treatments. As of February 25, 2017, 1209 patients had enrolled, the majority from Israel (44.3%) and the US (31.4%). Median age at GOS entry was 40.4 years, 44.1% were male, and 13.3% had undergone a total splenectomy. Most patients had type 1 GD (91.5%) and were of Ashkenazi Jewish ethnicity (55.8%). N370S/N370S was the most prevalent genotype, accounting for 44.2% of genotype-confirmed individuals (n = 847); however, there was considerable variation between countries. A total of 887 (73.4%) patients had received ≥1 GD-specific treatment at any time, most commonly imiglucerase (n = 587), velaglucerase alfa (n = 507), and alglucerase (n = 102). Hematological and visceral findings at the time of GOS entry were close to normal for most patients, probably a result of previous treatment; however, spleen volume of patients in Israel was almost double that of patients elsewhere (7.2 multiples of normal [MN] vs. 2.7, 2.9 and 4.9 MN in the US, UK and rest of world), which may be explained by a greater disease severity in this cohort. This analysis aimed to provide an overview of GOS and present baseline demographic and disease characteristics of participating patients to help improve the understanding of the natural history of GD and inform the overall management of patients with the disease.
© 2017 The Authors American Journal of Hematology Published by Wiley Periodicals, Inc.
Figures
References
-
- Zimran A, Elstein D, Gaucher disease and related lysosomal storage diseases In: Kaushansky K, Lichtman M, Prchal J, et al., eds. Williams Hematology. 9th ed New York, NY: McGraw‐Hill; 2016.
-
- Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83(1–2):6–15. - PubMed
-
- Nalysnyk L, Rotella P, Simeone JC, et al. Gaucher disease epidemiology and natural history: a comprehensive review of the literature. Hematology. 2017;22(2):65–73. - PubMed
-
- Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency–macrophage‐targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991;324(21):1464–1470. - PubMed
-
- Grabowski GA, Barton NW, Pastores G, et al. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose‐terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med. 1995;122(1):33–39. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials