

DINC 2.0: A New Protein-Peptide Docking Webserver Using an Incremental Approach
- PMID: 29092940
- PMCID: PMC5679007
- DOI: 10.1158/0008-5472.CAN-17-0511
DINC 2.0: A New Protein-Peptide Docking Webserver Using an Incremental Approach
Abstract
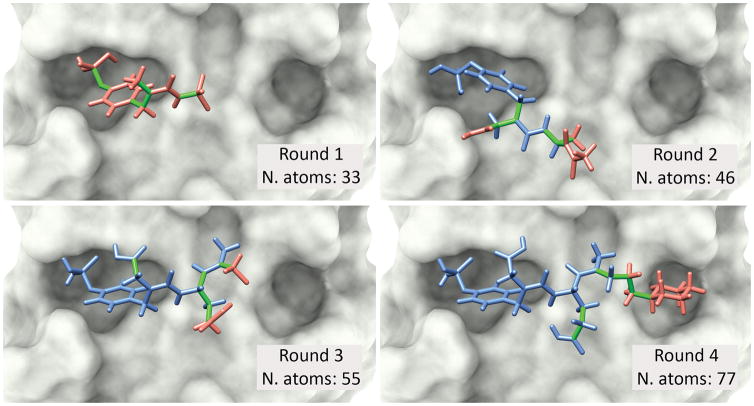
Molecular docking is a standard computational approach to predict binding modes of protein-ligand complexes by exploring alternative orientations and conformations of the ligand (i.e., by exploring ligand flexibility). Docking tools are largely used for virtual screening of small drug-like molecules, but their accuracy and efficiency greatly decays for ligands with more than 10 flexible bonds. This prevents a broader use of these tools to dock larger ligands, such as peptides, which are molecules of growing interest in cancer research. To overcome this limitation, our group has previously proposed a meta-docking strategy, called DINC, to predict binding modes of large ligands. By incrementally docking overlapping fragments of a ligand, DINC allowed predicting binding modes of peptide-based inhibitors of transcription factors involved in cancer. Here, we describe DINC 2.0, a revamped version of the DINC webserver with enhanced capabilities and a more user-friendly interface. DINC 2.0 allows docking ligands that were previously too challenging for DINC, such as peptides with more than 25 flexible bonds. The webserver is freely accessible at http://dinc.kavrakilab.org, together with additional documentation and video tutorials. Our team will provide continuous support for this tool and is working on extending its applicability to other challenging fields, such as personalized immunotherapy against cancer. Cancer Res; 77(21); e55-57. ©2017 AACR.
©2017 American Association for Cancer Research.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
