

Combating subclonal evolution of resistant cancer phenotypes
- PMID: 29093439
- PMCID: PMC5666005
- DOI: 10.1038/s41467-017-01174-3
Combating subclonal evolution of resistant cancer phenotypes
Erratum in
-
Publisher Correction: Combating subclonal evolution of resistant cancer phenotypes.Nat Commun. 2018 Feb 5;9(1):572. doi: 10.1038/s41467-017-02383-6. Nat Commun. 2018. PMID: 29402882 Free PMC article.
Abstract
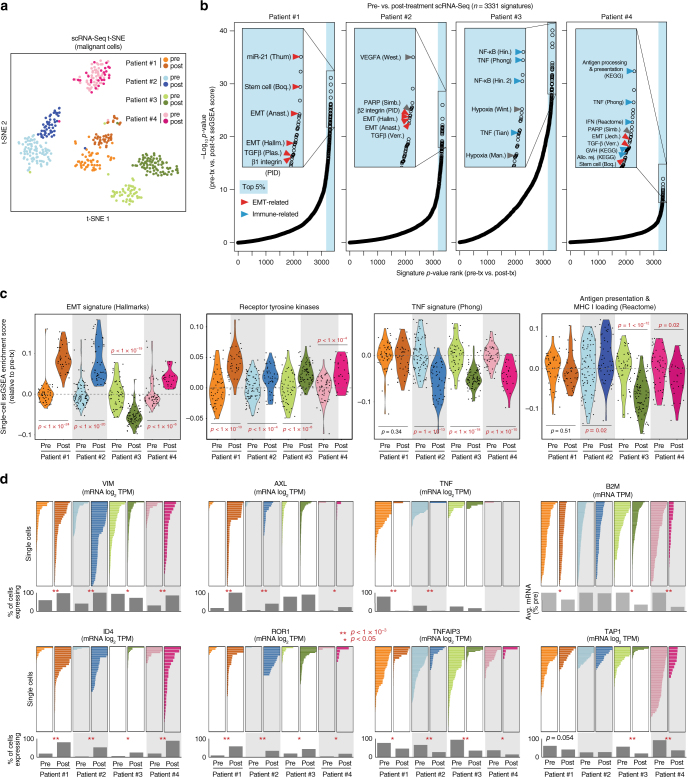
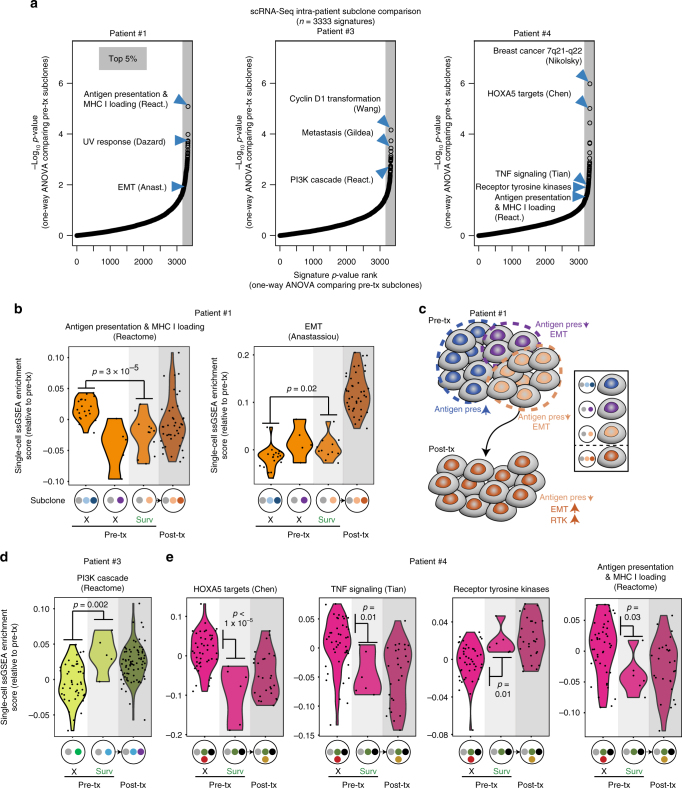
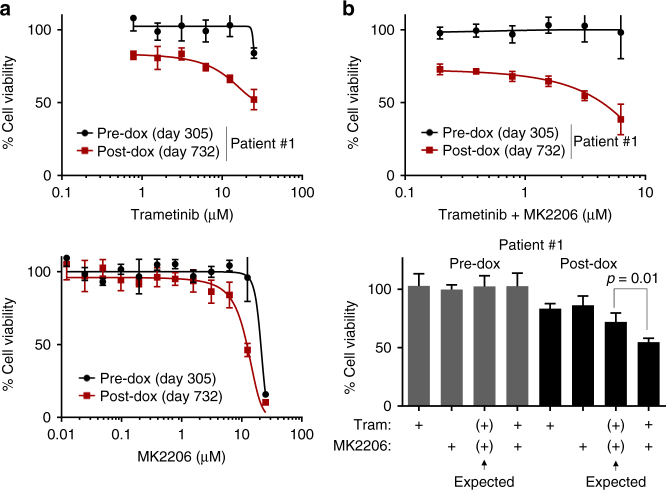
Metastatic breast cancer remains challenging to treat, and most patients ultimately progress on therapy. This acquired drug resistance is largely due to drug-refractory sub-populations (subclones) within heterogeneous tumors. Here, we track the genetic and phenotypic subclonal evolution of four breast cancers through years of treatment to better understand how breast cancers become drug-resistant. Recurrently appearing post-chemotherapy mutations are rare. However, bulk and single-cell RNA sequencing reveal acquisition of malignant phenotypes after treatment, including enhanced mesenchymal and growth factor signaling, which may promote drug resistance, and decreased antigen presentation and TNF-α signaling, which may enable immune system avoidance. Some of these phenotypes pre-exist in pre-treatment subclones that become dominant after chemotherapy, indicating selection for resistance phenotypes. Post-chemotherapy cancer cells are effectively treated with drugs targeting acquired phenotypes. These findings highlight cancer's ability to evolve phenotypically and suggest a phenotype-targeted treatment strategy that adapts to cancer as it evolves.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
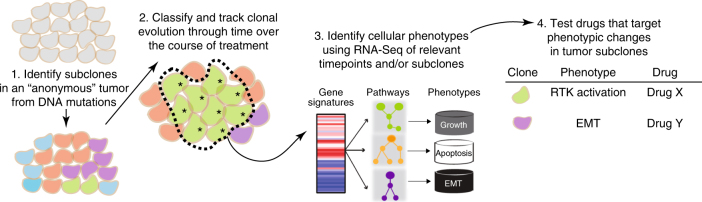
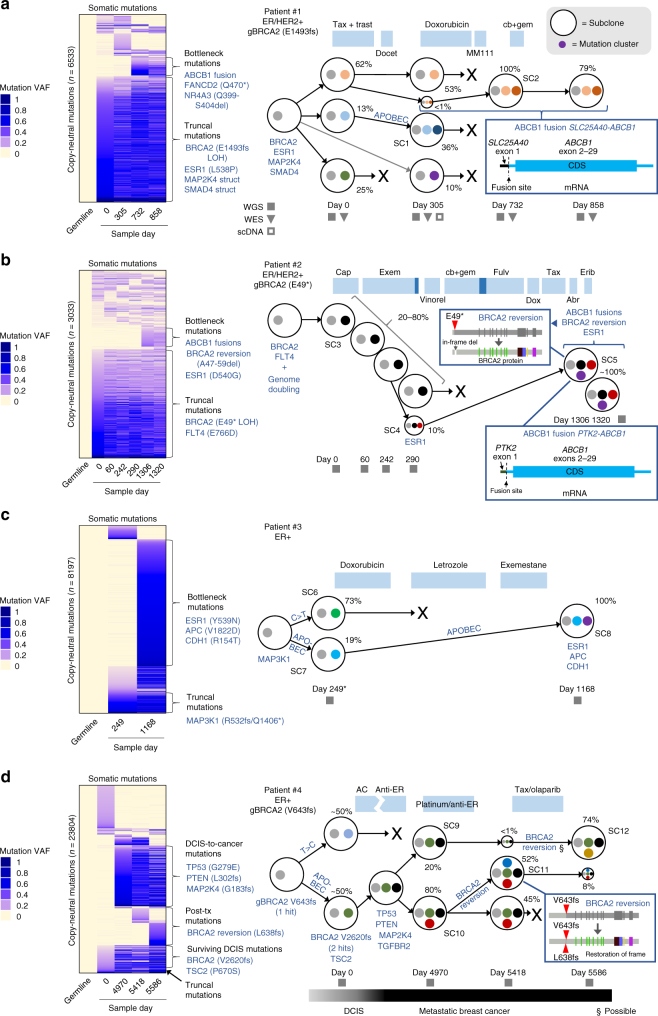
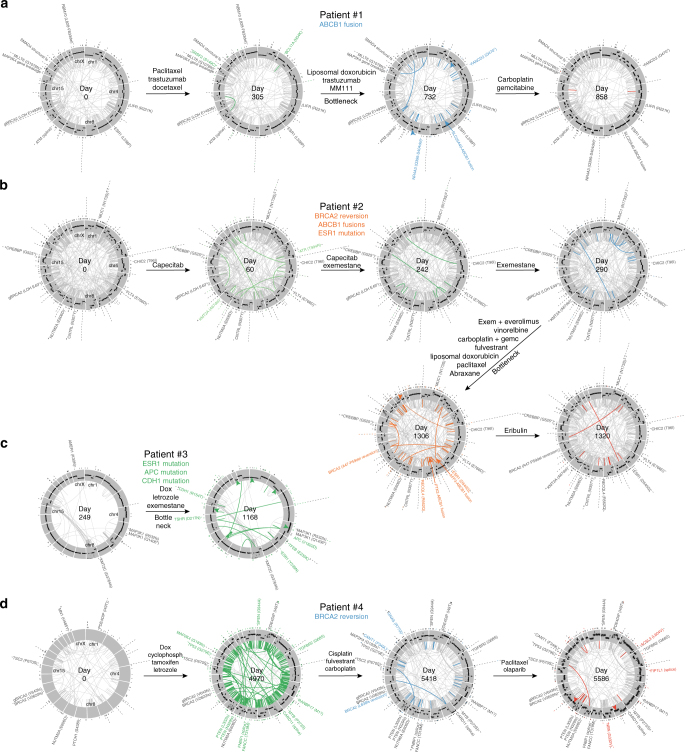
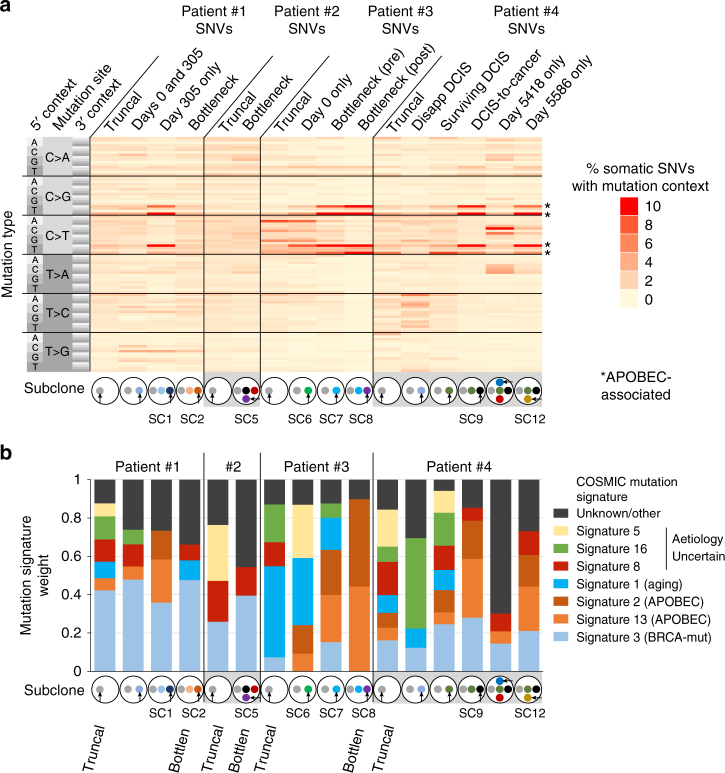
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
