

Two independent modes of chromatin organization revealed by cohesin removal
- PMID: 29094699
- PMCID: PMC5687303
- DOI: 10.1038/nature24281
Two independent modes of chromatin organization revealed by cohesin removal
Abstract
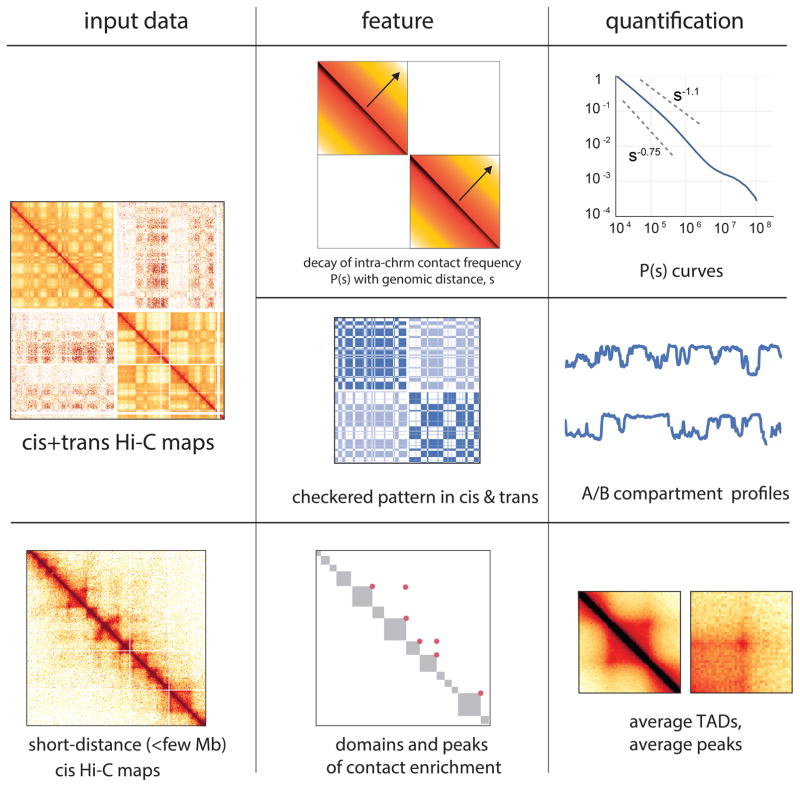
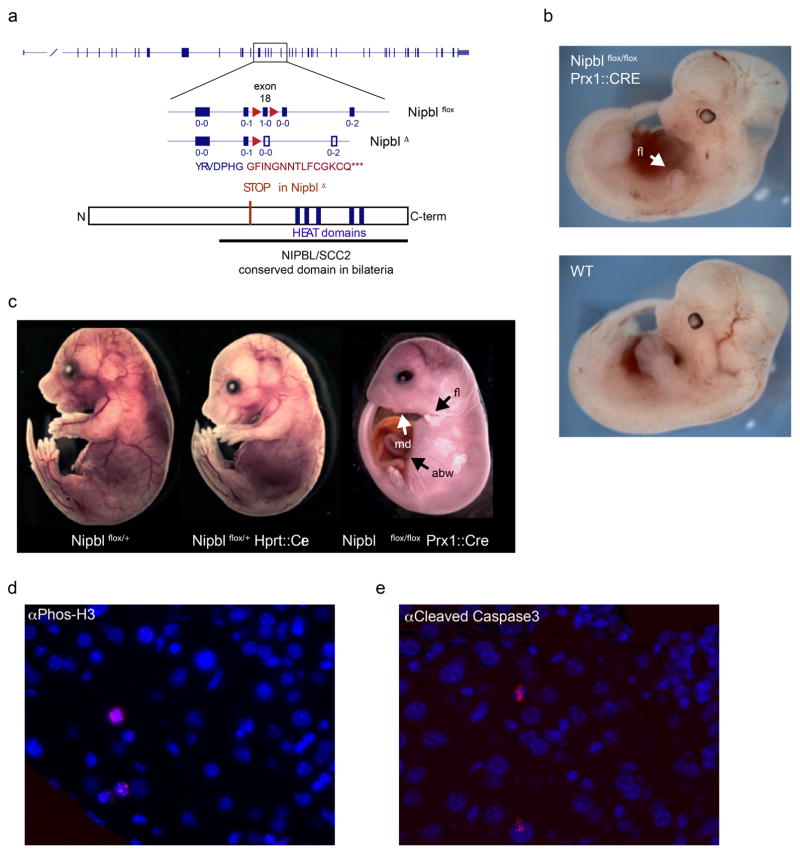
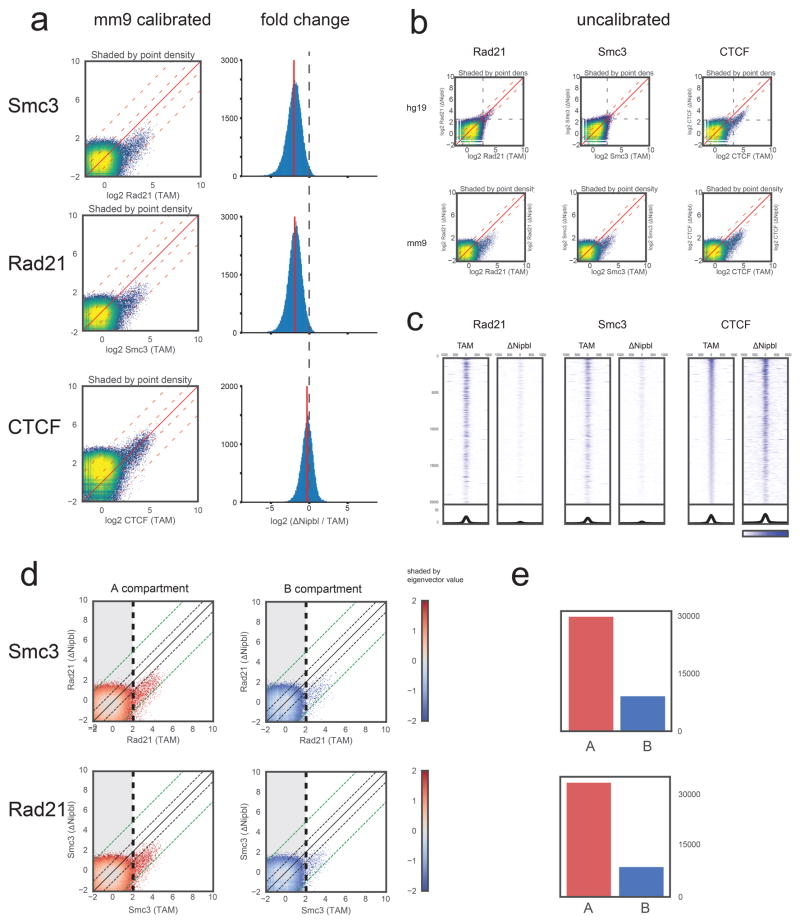
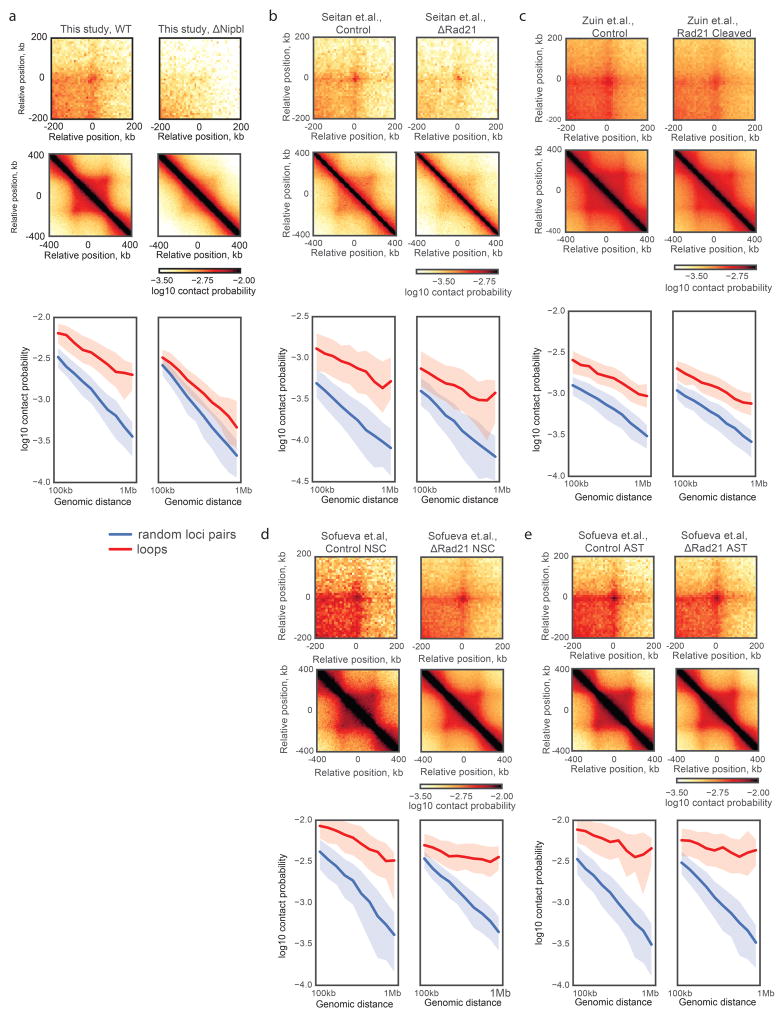
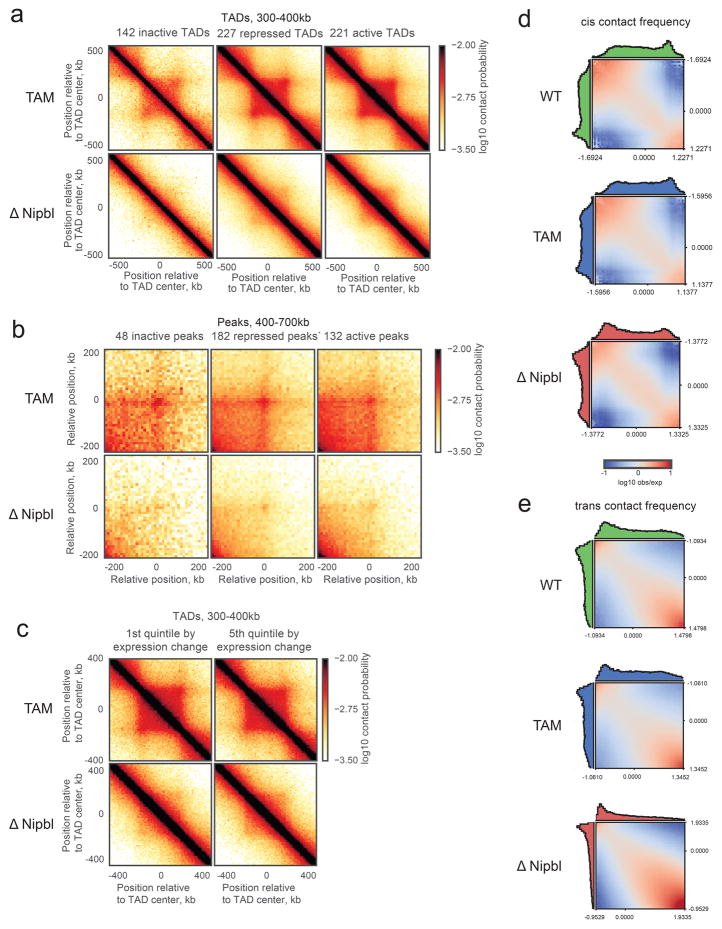
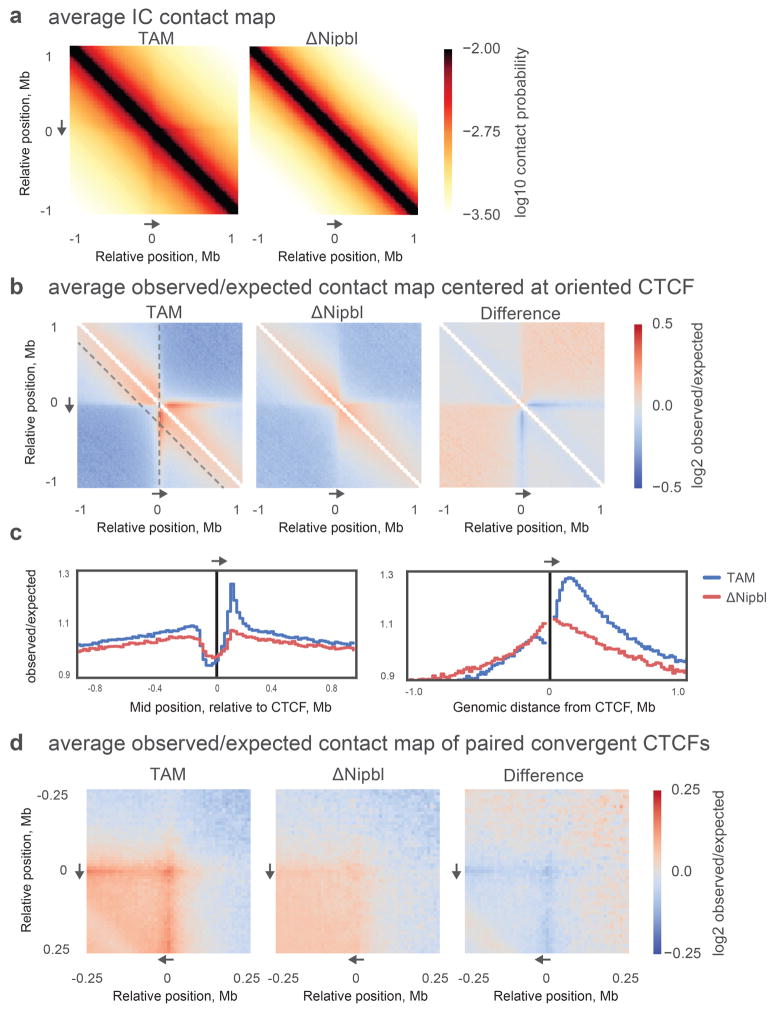
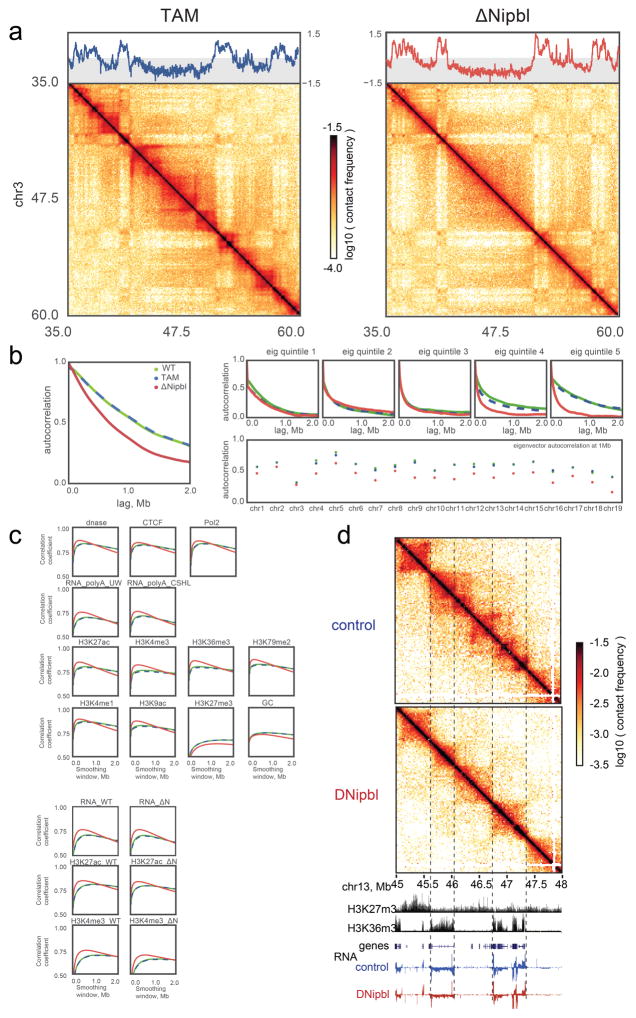
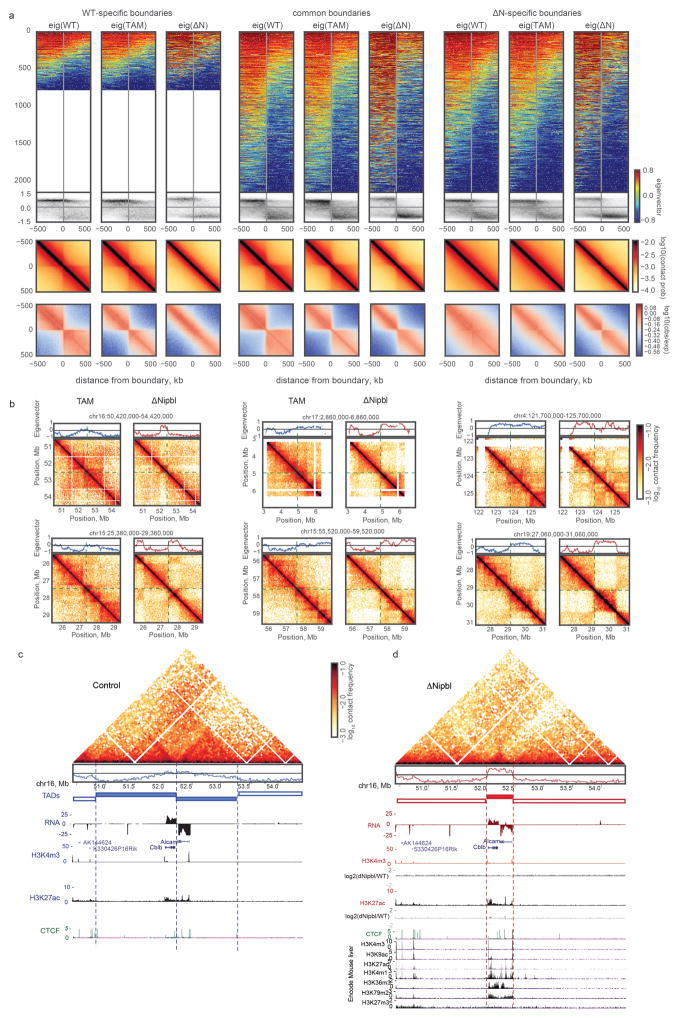
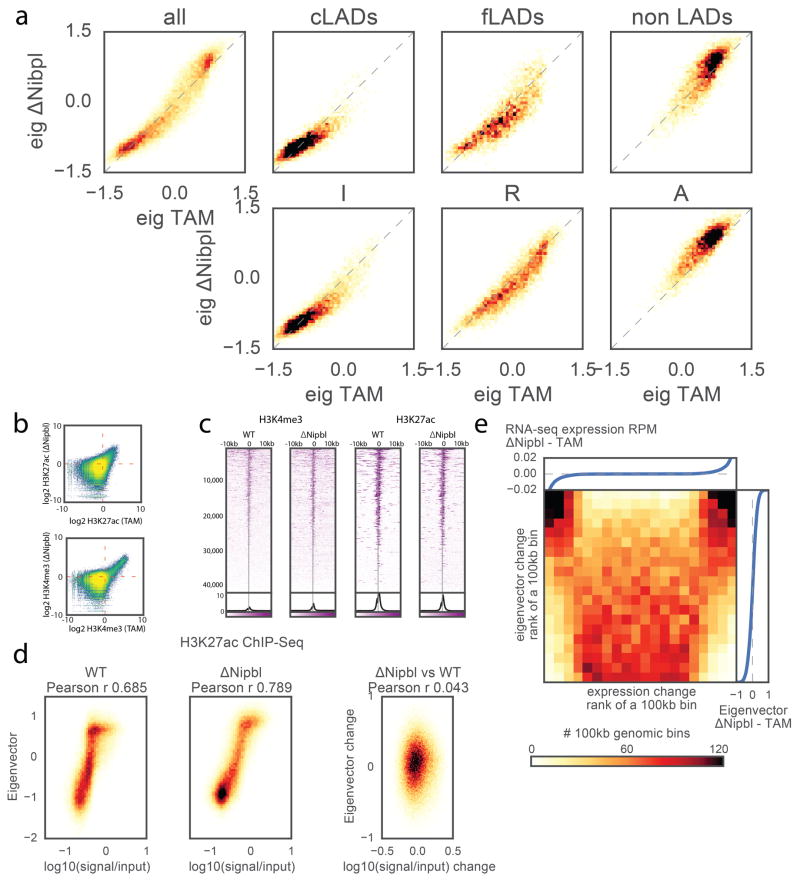
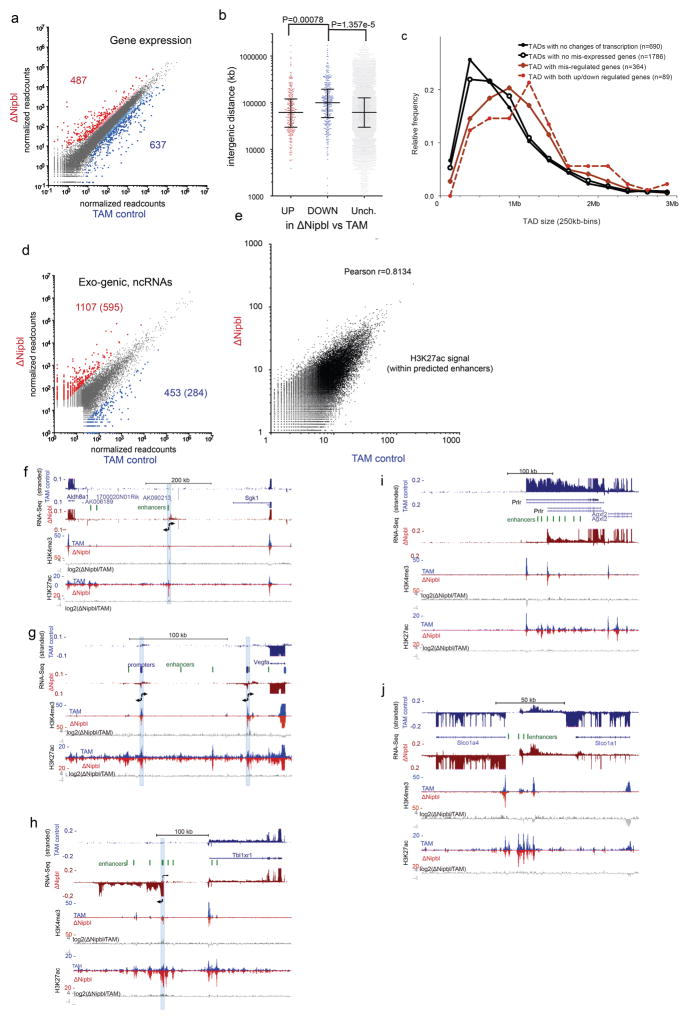
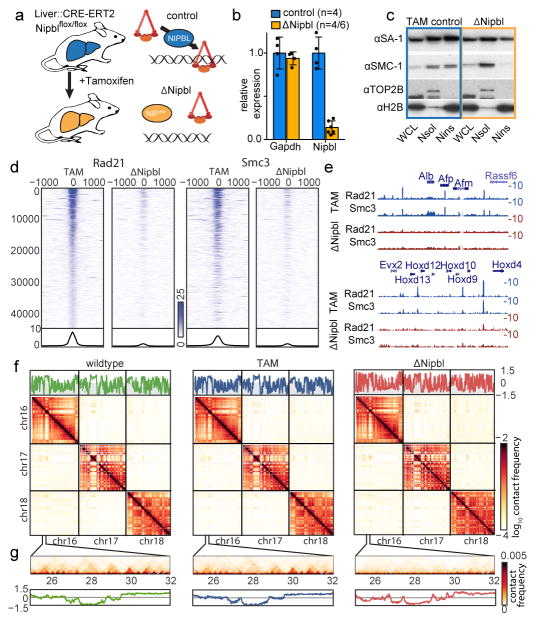
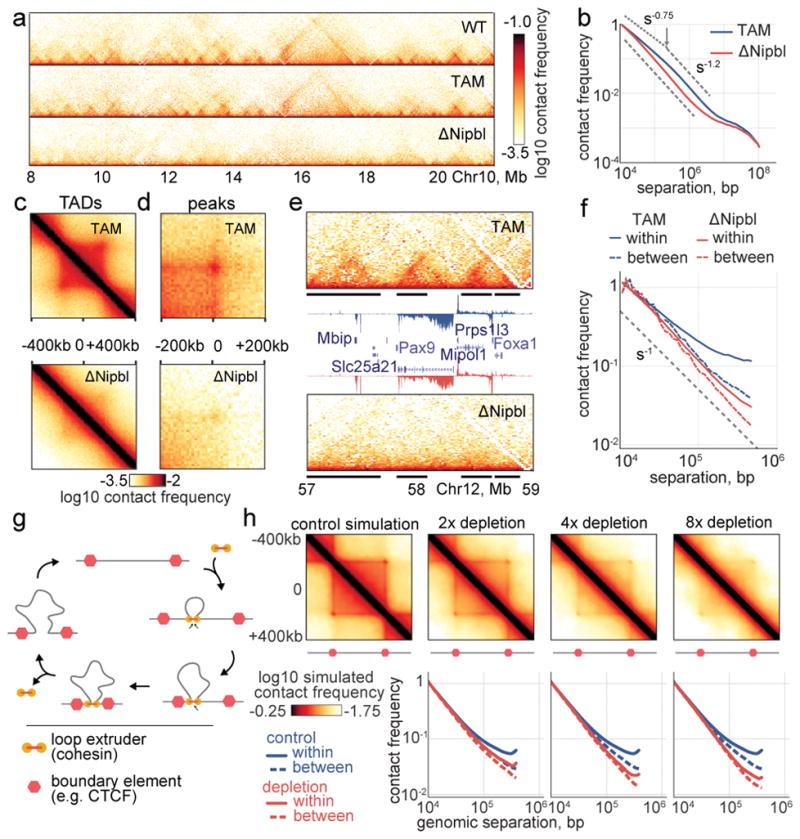
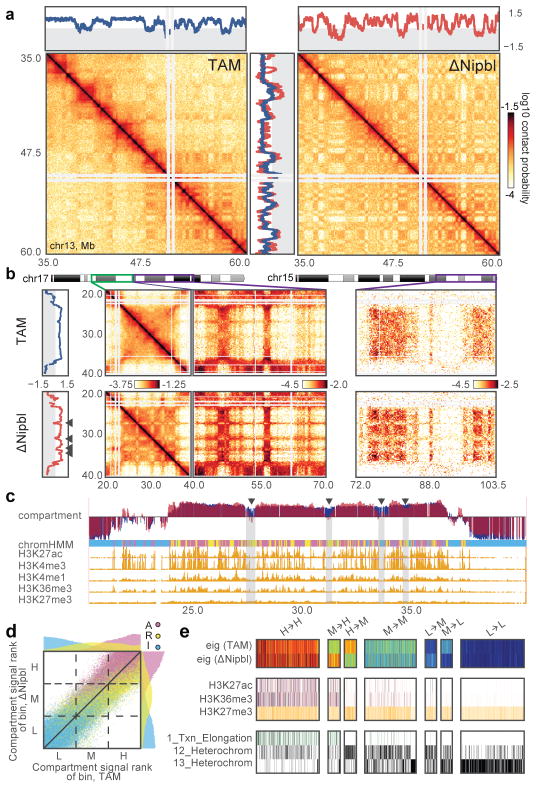
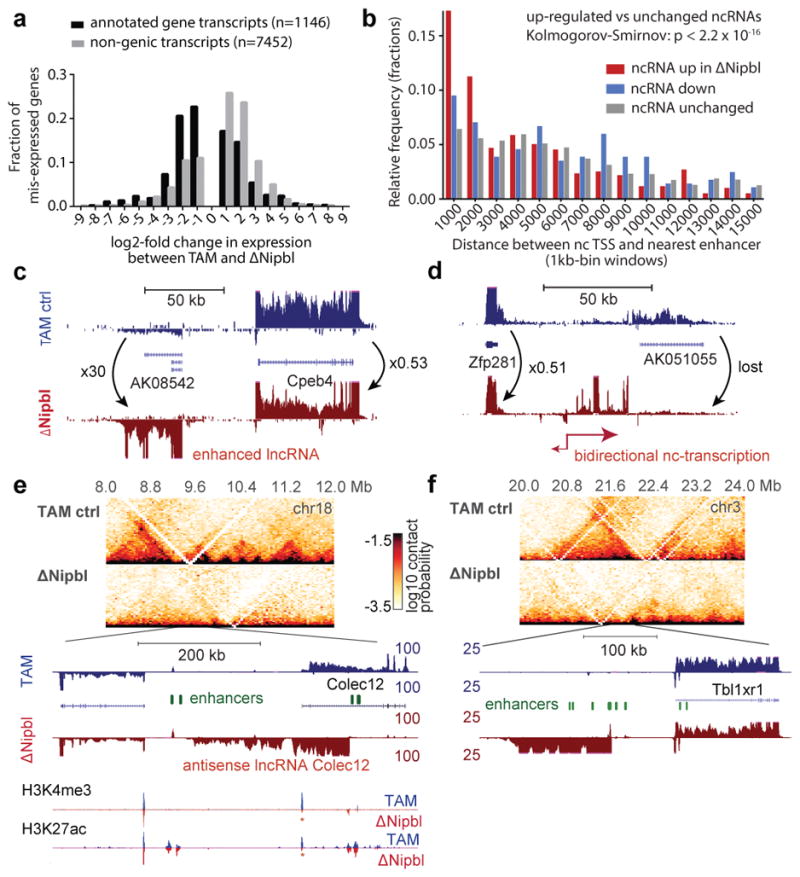
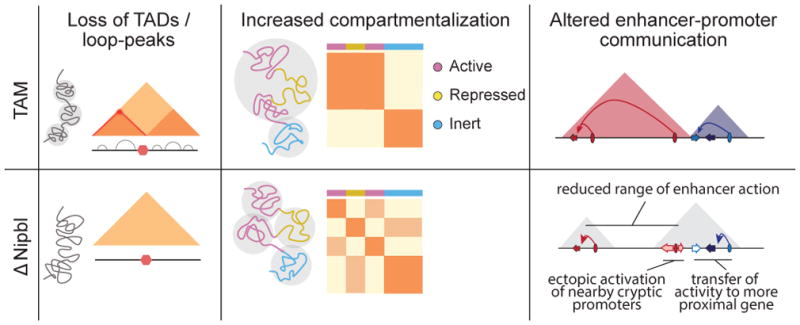
Imaging and chromosome conformation capture studies have revealed several layers of chromosome organization, including segregation into megabase-sized active and inactive compartments, and partitioning into sub-megabase domains (TADs). It remains unclear, however, how these layers of organization form, interact with one another and influence genome function. Here we show that deletion of the cohesin-loading factor Nipbl in mouse liver leads to a marked reorganization of chromosomal folding. TADs and associated Hi-C peaks vanish globally, even in the absence of transcriptional changes. By contrast, compartmental segregation is preserved and even reinforced. Strikingly, the disappearance of TADs unmasks a finer compartment structure that accurately reflects the underlying epigenetic landscape. These observations demonstrate that the three-dimensional organization of the genome results from the interplay of two independent mechanisms: cohesin-independent segregation of the genome into fine-scale compartments, defined by chromatin state; and cohesin-dependent formation of TADs, possibly by loop extrusion, which helps to guide distant enhancers to their target genes.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Chromosome biology: How to build a cohesive genome in 3D.Nature. 2017 Nov 2;551(7678):38-40. doi: 10.1038/nature24145. Epub 2017 Oct 4. Nature. 2017. PMID: 28976964 No abstract available.
-
Genome organization: Compartmentalizing chromatin without cohesin.Nat Rev Genet. 2017 Nov;18(11):640-641. doi: 10.1038/nrg.2017.84. Epub 2017 Oct 9. Nat Rev Genet. 2017. PMID: 28989172 No abstract available.
References
-
- Bickmore WA, van Steensel B. Genome Architecture: Domain Organization of Interphase Chromosomes. Cell. 2013;152:1270–1284. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
