

Optimisation of 16S rRNA gut microbiota profiling of extremely low birth weight infants
- PMID: 29096601
- PMCID: PMC5668952
- DOI: 10.1186/s12864-017-4229-x
Optimisation of 16S rRNA gut microbiota profiling of extremely low birth weight infants
Abstract
Background: Infants born prematurely, particularly extremely low birth weight infants (ELBW) have altered gut microbial communities. Factors such as maternal health, gut immaturity, delivery mode, and antibiotic treatments are associated with microbiota disturbances, and are linked to an increased risk of certain diseases such as necrotising enterocolitis. Therefore, there is a requirement to optimally characterise microbial profiles in this at-risk cohort, via standardisation of methods, particularly for studying the influence of microbiota therapies (e.g. probiotic supplementation) on community profiles and health outcomes. Profiling of faecal samples using the 16S rRNA gene is a cost-efficient method for large-scale clinical studies to gain insights into the gut microbiota and additionally allows characterisation of cohorts were sample quantities are compromised (e.g. ELBW infants). However, DNA extraction method, and the 16S rRNA region targeted can significantly change bacterial community profiles obtained, and so confound comparisons between studies. Thus, we sought to optimise a 16S rRNA profiling protocol to allow standardisation for studying ELBW infant faecal samples, with or without probiotic supplementation.
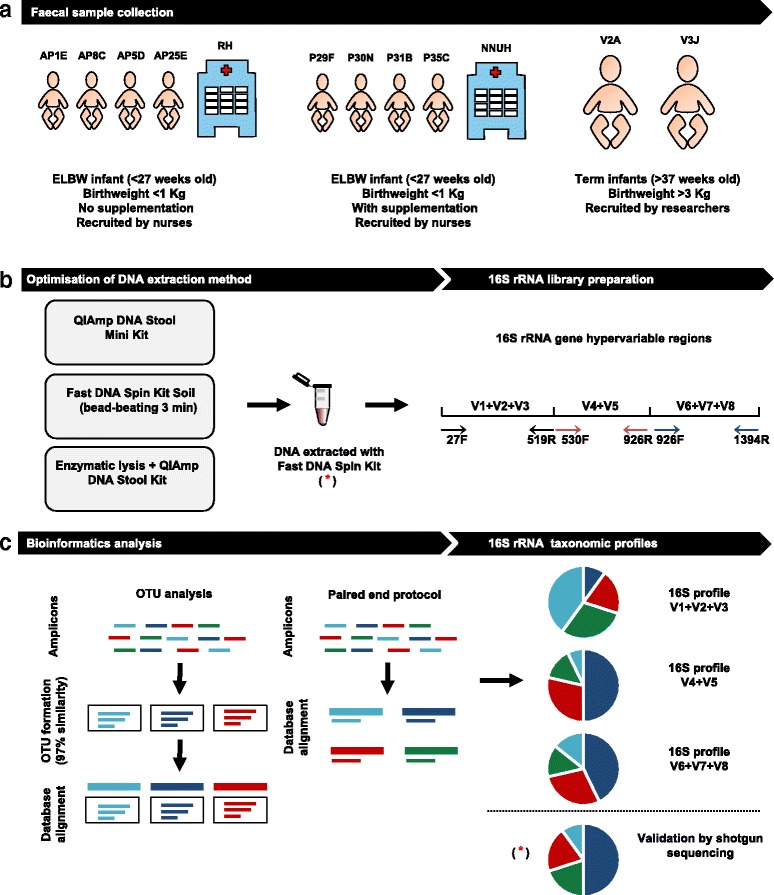
Methods: Using ELBW faecal samples, we compared three different DNA extraction methods, and subsequently PCR amplified and sequenced three hypervariable regions of the 16S rRNA gene (V1 + V2 + V3), (V4 + V5) and (V6 + V7 + V8), and compared two bioinformatics approaches to analyse results (OTU and paired end). Paired shotgun metagenomics was used as a 'gold-standard'.
Results: Results indicated a longer bead-beating step was required for optimal bacterial DNA extraction and that sequencing regions (V1 + V2 + V3) and (V6 + V7 + V8) provided the most representative taxonomic profiles, which was confirmed via shotgun analysis. Samples sequenced using the (V4 + V5) region were found to be underrepresented in specific taxa including Bifidobacterium, and had altered diversity profiles. Both bioinformatics 16S rRNA pipelines used in this study (OTU and paired end) presented similar taxonomic profiles at genus level.
Conclusions: We determined that DNA extraction from ELBW faecal samples, particularly those infants receiving probiotic supplementation, should include a prolonged beat-beating step. Furthermore, use of the 16S rRNA (V1 + V2 + V3) and (V6 + V7 + V8) regions provides reliable representation of ELBW microbiota profiles, while inclusion of the (V4 + V5) region may not be appropriate for studies where Bifidobacterium constitutes a resident microbiota member.
Keywords: 16S rRNA gene sequencing; Bifidobacterium; DNA extraction; Extremely low birth weight infants; Microbiota; Shotgun sequencing.
Conflict of interest statement
Ethics approval and consent to participate
Study was approved by the University of East Anglia (UEA) Faculty of Medical and Health Sciences Ethics Committee, and sample collection was in accordance with protocols laid out by the NRES approved UEA Biorepository (Licence no: 11,208). Parents gave written informed consent for their infant to participate in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures





References
-
- WHO. November 2016. http://www.who.int/mediacentre/factsheets/fs363/en/. Accessed 2 Mar 2017.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
