

GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly
- PMID: 29097403
- PMCID: PMC5741059
- DOI: 10.1101/gr.222109.117
GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly
Abstract
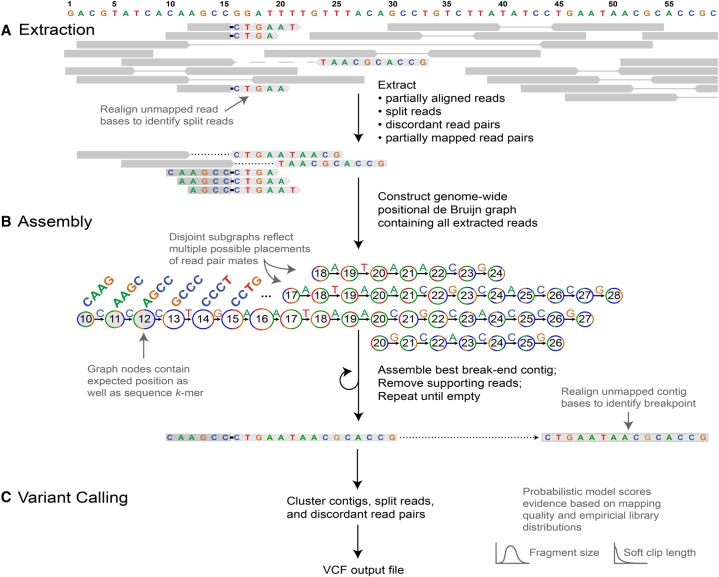
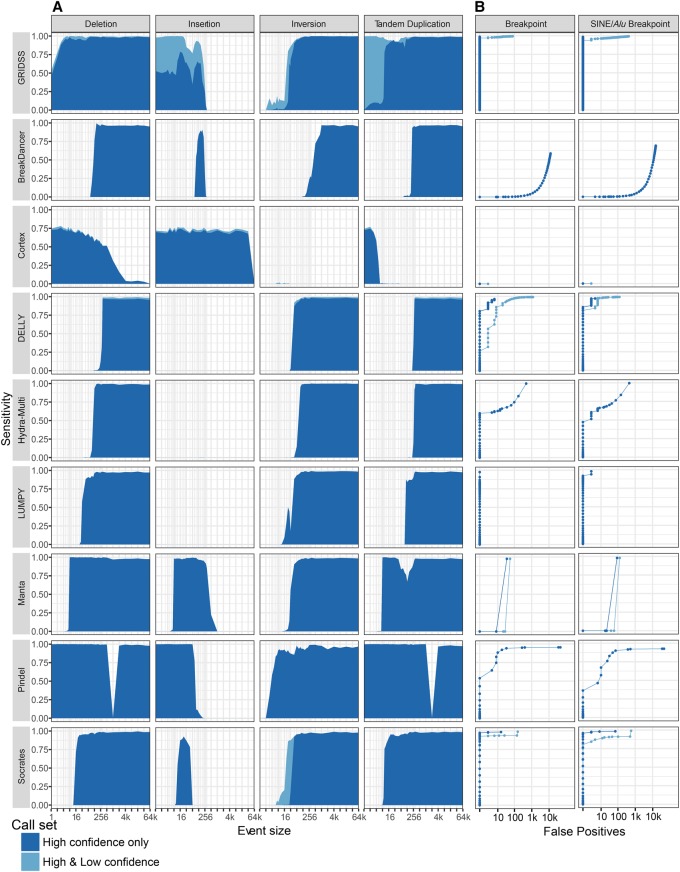
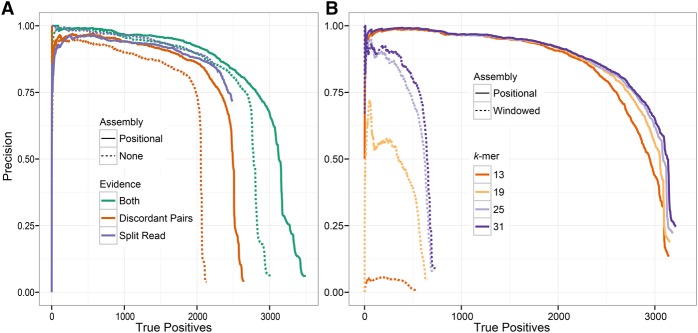
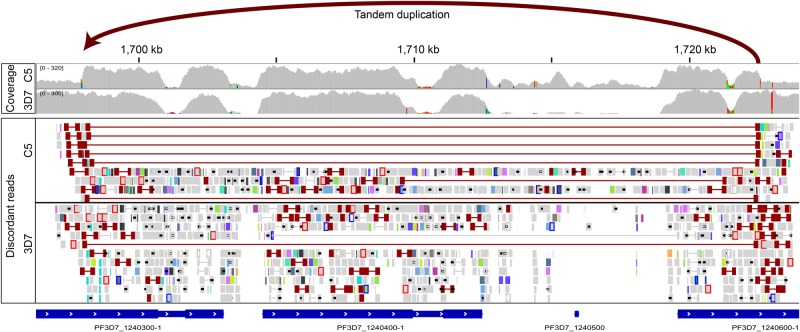
The identification of genomic rearrangements with high sensitivity and specificity using massively parallel sequencing remains a major challenge, particularly in precision medicine and cancer research. Here, we describe a new method for detecting rearrangements, GRIDSS (Genome Rearrangement IDentification Software Suite). GRIDSS is a multithreaded structural variant (SV) caller that performs efficient genome-wide break-end assembly prior to variant calling using a novel positional de Bruijn graph-based assembler. By combining assembly, split read, and read pair evidence using a probabilistic scoring, GRIDSS achieves high sensitivity and specificity on simulated, cell line, and patient tumor data, recently winning SV subchallenge #5 of the ICGC-TCGA DREAM8.5 Somatic Mutation Calling Challenge. On human cell line data, GRIDSS halves the false discovery rate compared to other recent methods while matching or exceeding their sensitivity. GRIDSS identifies nontemplate sequence insertions, microhomologies, and large imperfect homologies, estimates a quality score for each breakpoint, stratifies calls into high or low confidence, and supports multisample analysis.
© 2017 Cameron et al.; Published by Cold Spring Harbor Laboratory Press.
Figures

References
-
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, Cox AJ, Kruglyak S, Saunders CT. 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources