

Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing
- PMID: 29097521
- PMCID: PMC5829300
- DOI: 10.1126/science.aao0464
Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing
Abstract
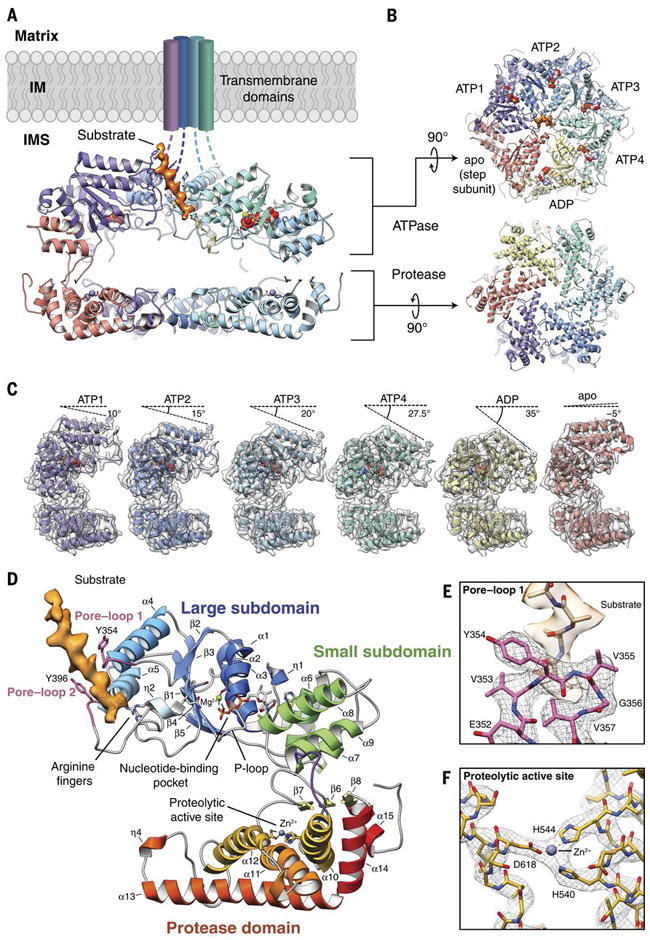
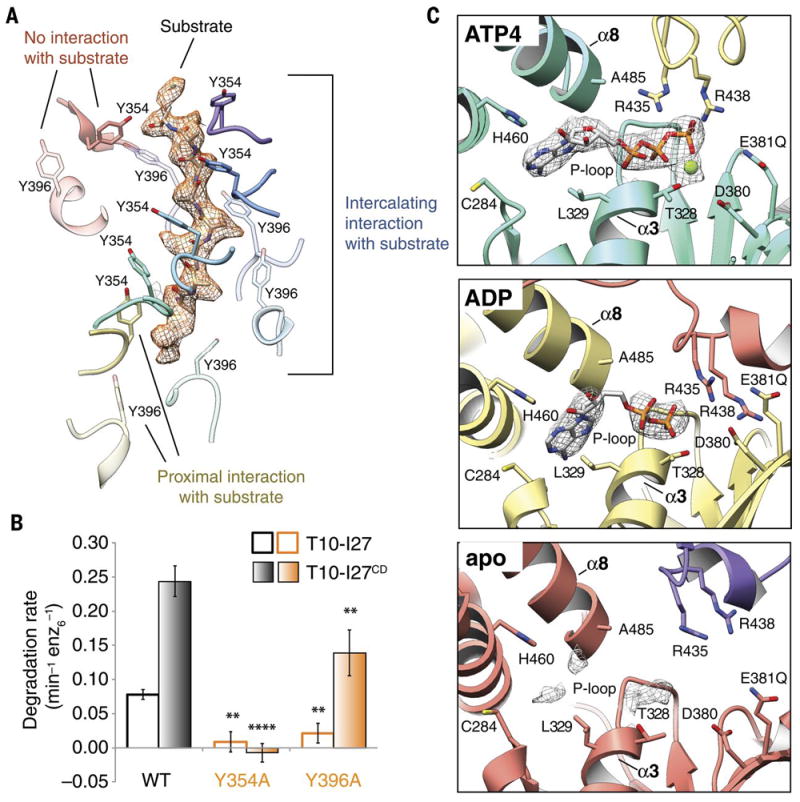
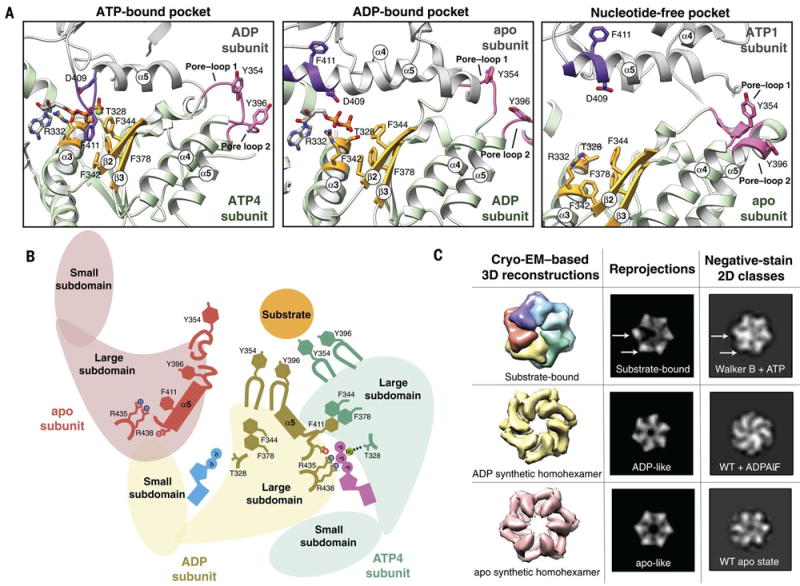
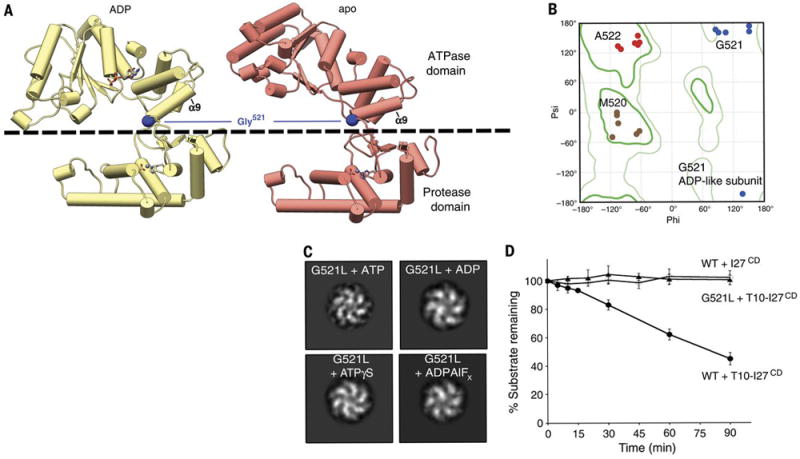
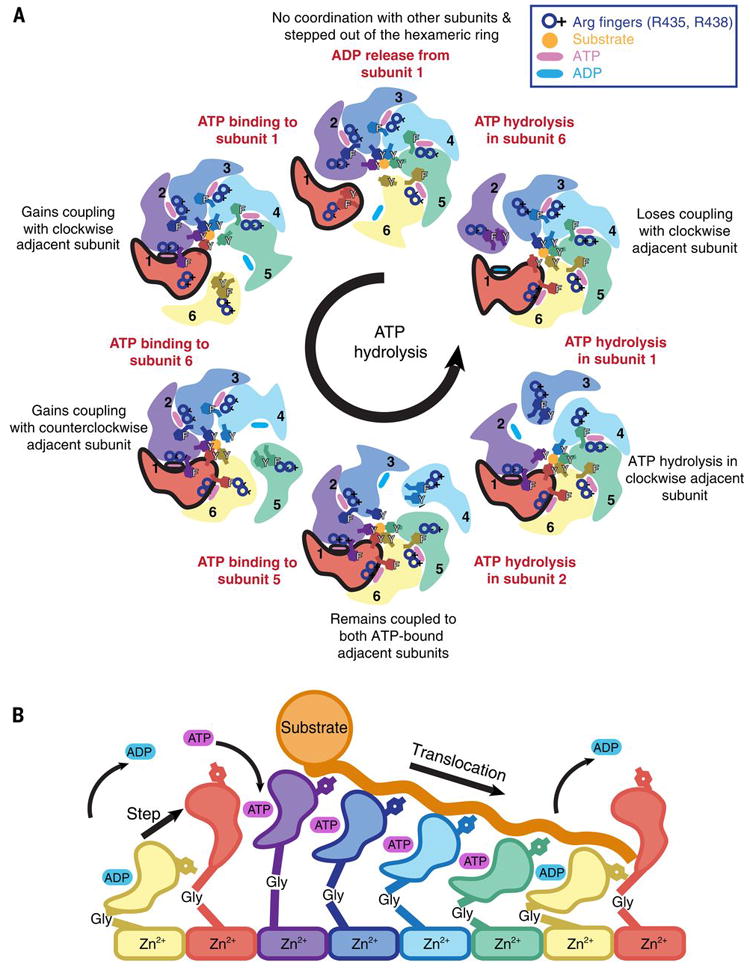
We present an atomic model of a substrate-bound inner mitochondrial membrane AAA+ quality control protease in yeast, YME1. Our ~3.4-angstrom cryo-electron microscopy structure reveals how the adenosine triphosphatases (ATPases) form a closed spiral staircase encircling an unfolded substrate, directing it toward the flat, symmetric protease ring. Three coexisting nucleotide states allosterically induce distinct positioning of tyrosines in the central channel, resulting in substrate engagement and translocation to the negatively charged proteolytic chamber. This tight coordination by a network of conserved residues defines a sequential, around-the-ring adenosine triphosphate hydrolysis cycle that results in stepwise substrate translocation. A hingelike linker accommodates the large-scale nucleotide-driven motions of the ATPase spiral relative to the planar proteolytic base. The translocation mechanism is likely conserved for other AAA+ ATPases.
Copyright © 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
References
-
- Quiros PM, Langer T, LopezOtin C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015;16:345–359. - PubMed
-
- Nolden M, et al. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–289. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
