

Viewing BCL2 and cell death control from an evolutionary perspective
- PMID: 29099481
- PMCID: PMC5729521
- DOI: 10.1038/cdd.2017.145
Viewing BCL2 and cell death control from an evolutionary perspective
Abstract
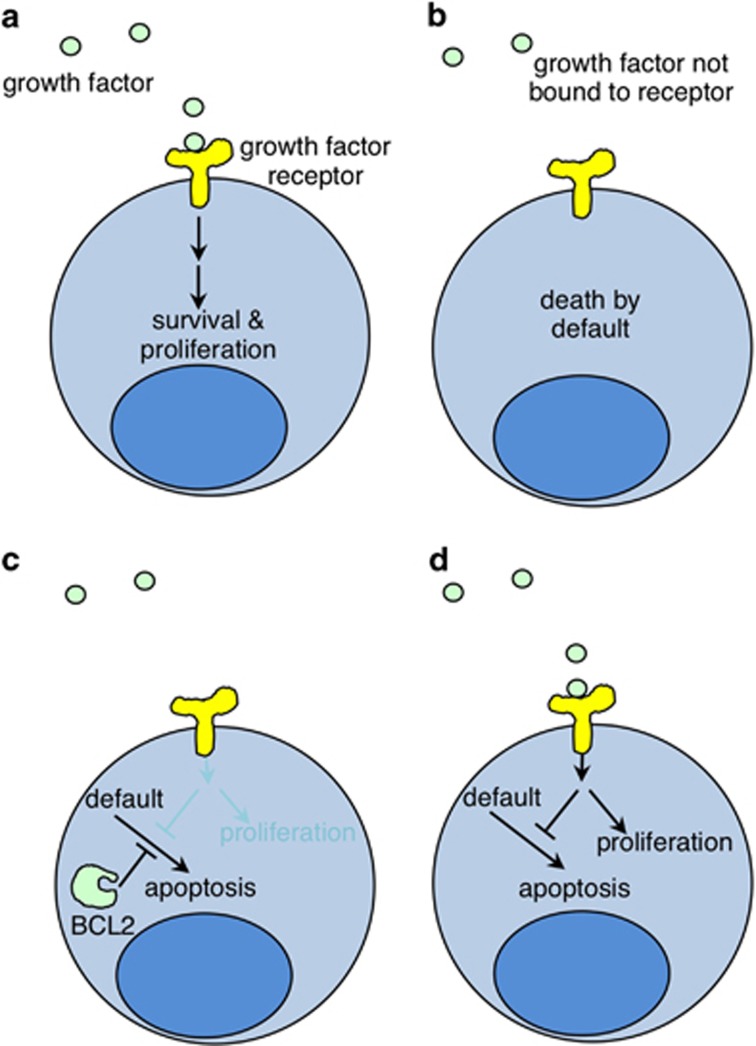
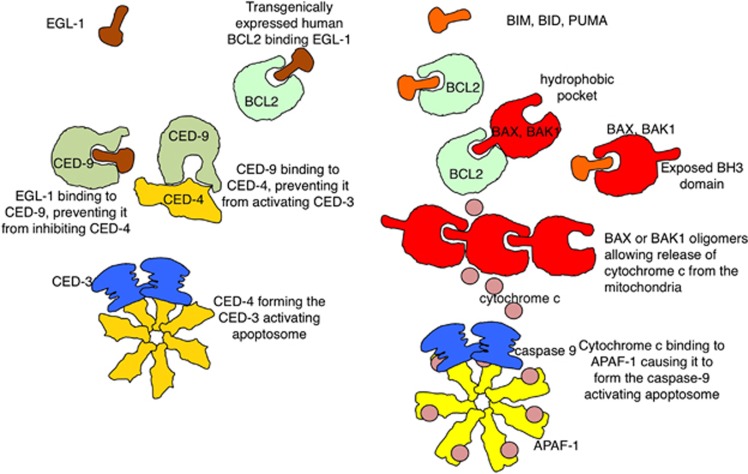
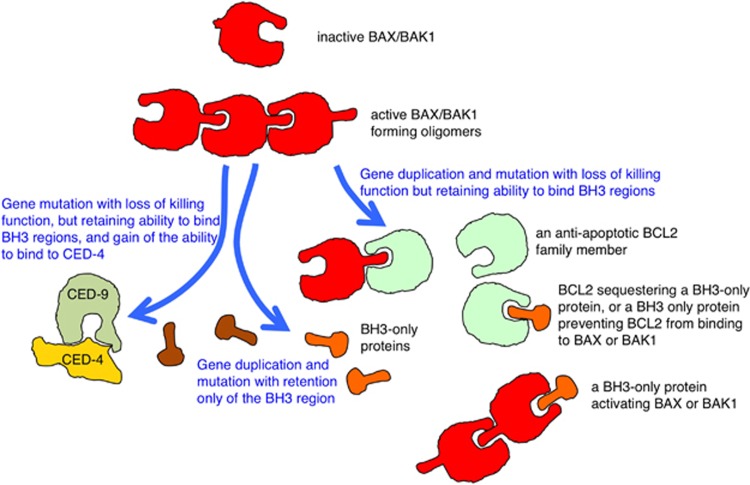
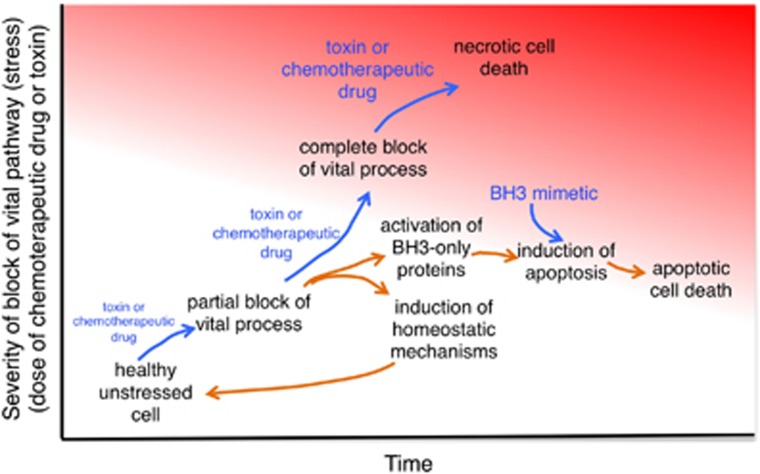
The last 30 years of studying BCL2 have brought cell death research into the molecular era, and revealed its relevance to human pathophysiology. Most, if not all metazoans use an evolutionarily conserved process for cellular self destruction that is controlled and implemented by proteins related to BCL2. We propose the anti-apoptotic BCL2-like and pro-apoptotic BH3-only members of the family arose through duplication and modification of genes for the pro-apoptotic multi-BH domain family members, such as BAX and BAK1. In that way, a cell suicide process that initially evolved as a mechanism for defense against intracellular parasites was then also used in multicellular organisms for morphogenesis and to maintain the correct number of cells in adults by balancing cell production by mitosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Fukuhara S, Rowley JD. Chromosome 14 translocations in non-Burkitt lymphomas. Int J Cancer 1978; 22: 14–21. - PubMed
-
- Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984; 226: 1097–1099. - PubMed
-
- Tsujimoto Y, Jaffe E, Cossman J, Gorham J, Nowell PC, Croce CM. Clustering of breakpoints on chromosome 11 in human B-cell neoplasms with the t(11;14) chromosome translocation. Nature 1985; 315: 340–343. - PubMed
-
- Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell 1986; 47: 19–28. - PubMed
-
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988; 335: 440–442. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
